
Academic Editor: Graham Pawelec
CAR (Chimeric antigen receptor)-T cell therapy has become a very promising type of immunotherapy against hematological cancers. This report is focused on CAR-T cells targeting immune checkpoint proteins expressed on tumor cells. The CD70, CD47, CD80, CD86, B7H3, B7H4, PDL-1, TIGIT CAR-T cells and other CAR-T cells are discussed as an effective approach to deplete tumor cells expressing checkpoint proteins. CAR-T cell therapy targeting checkpoint pathways is a promising therapy to decrease inhibitory signaling pathways. The review highlights future directions and perspectives in CAR-T cells targeting immune checkpoint pathways.
CAR-T cell therapy is a therapy using antibody-based chimeric antigen receptor expressing T cells targeting cell surface proteins on tumor cells. CAR-T cell therapy is an effective approach against hematological and solid cancers [1, 2, 3, 4, 5, 6]. CAR-T cell or T body cell was first described by Zelig Eshhar and his colleagues at the Weizmann Institute of Science in Israel [7, 8, 9, 10, 11, 12]. Several CAR-T cells targeting CD19 and BCMA for hematological cancers were recently approved by FDA for usage in clinic [13]. CAR includes a single chain variable fragment (ScFv) of antibody which binds tumor antigen [14]. CAR-T cells transduce anti-tumor activity through costimulatory domain and activation domain signaling [3, 15, 16].
Once CAR-T cell binds to a tumor antigen, the CAR-T cell proliferation and expansion is activated, turning on CAR-T cell cytotoxic functions and signaling accompanied with secretion of cytokines, granzymes causing tumor cell death [5, 12]. There are several challenges for CAR-T cell use against solid tumors such as repressive microenvironment, exhaustion of CAR-T cells, upregulation of inhibitory checkpoint pathways and delivery of CAR-T cells to the tumor site. This review describes CAR-T cells targeting checkpoint pathway molecules to effectively kill tumors.
There are many antigens expressed in hematological cancers which are targeted by CAR-T cells such as CD19, CD22, CD37, CD20, CD33, BCMA [6] and in solid cancers - mesothelin, EGFR (Epidermal growth factor receptor), Her-2 (human epidermal growth factor receptor 2), PSMA (prostate-specific membrane antigen), CD133, CD47, ROR-1 (receptor tyrosine kinase-like orphan receptor 1), MUC-1 (mucin 1), CEA (carcinoembryonic antigen) [17] and other [1]. Many tumors overexpress immune checkpoint protein players such as PD-L1, CD112, CD155, B7H3, B7H4, CD80, CD86 affecting immune response signaling as reviewed in [18, 19]. In this review we will describe main checkpoint proteins and specifically focus on CAR-T cells targeting these checkpoint proteins.
We will also highlight several CAR-T cells targeting checkpoint molecules on other types of immune cells such as macrophages, Treg or NK cells. We will provide future directions for the field.
The CAR structure includes ScFv, hinge, transmembrane, co-stimulatory (CD28, CD137 (4-1BB), and activation domain such as CD3 zeta (Fig. 1). The ScFv consists of variable light (VL) and heavy (VH) chains of antibody fused in frame with the linker either in VL-linker-VH or VH-linker-VL format. The first generation of CAR had one CD3 domain; the second generation of CAR had two costimulatory domains and one CD3 activation domain; and the third generation of CAR had two costimulatory domains and CD3 zeta activation domain, as shown in Fig. 1.
 Fig. 1.
Fig. 1.CAR-T cell structures. First, second, third, fourth and fifth generation of CARs. ScFv, single chain variable fragment; VH-variable heavy chain; VL, variable light chain, hinge, transmembrane, CD28, 4-1BB or other costimulatory domains, CD3 zeta activation domain. The first generation of CAR included one CD3 zeta activation domain; the second generation of CAR included one costimulatory domain and one CD3 zeta activation domain. The third generation of CAR includes two costimulatory domains (for example, CD28 and 4-1BB) and one activation CD3 zeta domain. The fourth generation of CAR included second generation of CAR with secreted cytokine or chemokine protein. The fifth generation of CAR included additional expression of receptor domain for example IL-2 receptor chain beta stimulating STAT3/STAT5 signaling.
The fourth generation of CAR includes addition of secreted or membrane-bound
cytokines such as IL-15, IL-12, IL-18, IL-21, and IL-23 using either T2A, F2A,
P2A self-cleaving peptides, or adding IRES (internal ribosome entry site), using
a separate promoter [20, 21, 22, 23] or using gene-editing technology to insert additional
cargo players [24]. These CAR-T cells can express cytokine proteins such as
IL-12. The fifth generation CAR-T cells includes additional intracellular domain
of cytokine receptors such as IL-2 receptor beta chain (IL-2 R
The future designs of CAR will include CARs with more safety and higher efficacy. For example, CAR-T cells can secrete bispecific antibody to increase their efficacy by targeting tumor and tumor microenvironment [27, 28]. One elegant study used CAR-T cells secreting BITE bispecific antibody targeting fibroblast activation protein alpha, FAP with pro-tumorigenic role in tumor microenvironment [28].
One of the challenges in targeting solid tumors is on-target off-tumor toxicity due to expression of tumor antigens at low levels in normal tissues [29]. Thus, different “off switches” can be introduced to the CAR structure in order to deplete CAR-T cells in case of toxicity. The CAR structure can include different safety switches such as inducible caspase-9 suicide switch [30, 31], truncated EGFR (t-EGFR), RQR8 [32] or other switches to increase CAR-T cell safety. As an example, the inducible caspase-9 switch can eliminate majority of CAR-T cells within 30 minutes providing increased safety in clinic [33]. The switches are important for decreasing on-target off-tumor potential toxic effects in clinical studies.
T cells receive either costimulatory or inhibiting immune signals through interaction of immune receptors and ligands, which are called immune checkpoints. The costimulatory or inhibitory checkpoint signaling interaction and pathways between tumor and immune cells are shown on Fig. 2. The main costimulatory immune checkpoints include: CD80, CD86 ligands and CD28 receptor; CD70 ligand and CD27 receptor; OX40 ligand and OX40 receptor; CD137 or 41BB ligand and CD137 (4-1BB) receptor, ICOS ligand and ICOS receptors (Fig. 2, left panel). The main inhibitory immune checkpoints are VISTA; B7H3; B7H4 ligands/receptors; PD-L1/PD-L2/PD-1; CD80, CD86/CTLA-4; CD112/CD155/TIGIT; Gal-9/TIM3; MHC-peptide/LAG-3/KIR (Fig. 2). The different antibodies targeting inhibitory checkpoint pathways were developed in clinic such as FDA-approved Pembrolizumab (Keytruda), Nivolumab (Opdivo) targeting PD-1; ipilimumab (Yervoy) targeting CTLA-4; Avelumab (Bavencio), Atezolizumab (Tecentriq) targeting PD-L1. Tumors use the checkpoint pathways to avoid autoimmunity during normal conditions [34].
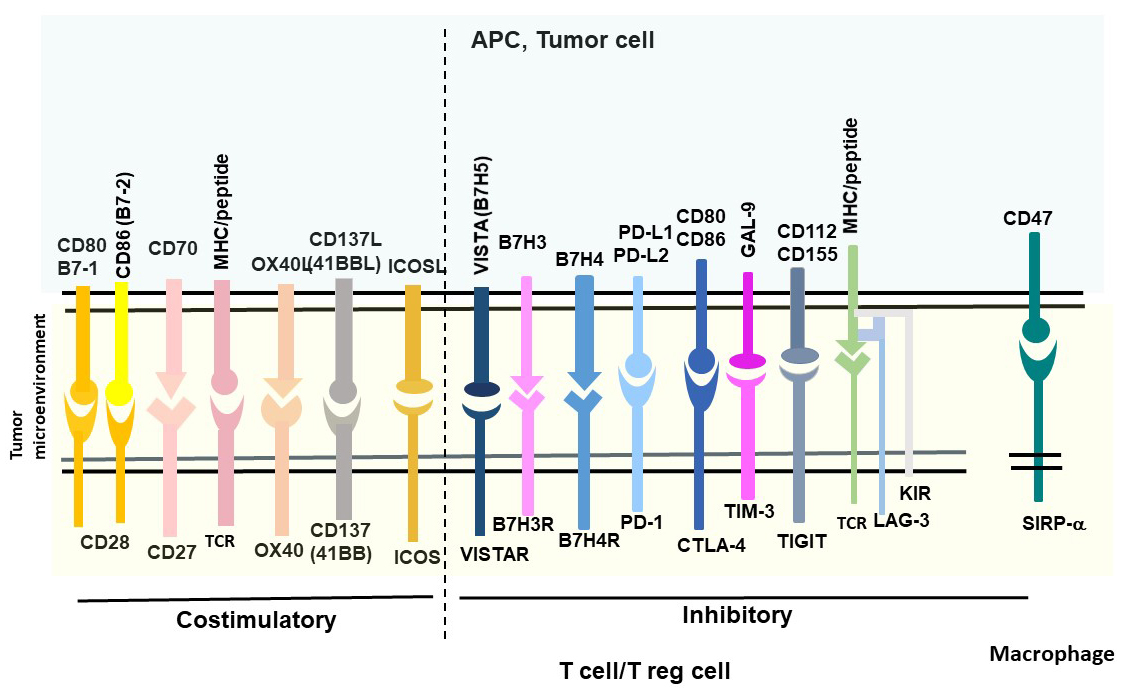 Fig. 2.
Fig. 2.Costimulatory and inhibitory checkpoint pathways. T/Treg cells and macrophages are immune cells with checkpoint receptors, and tumor cells express ligands.
The tumor microenvironment (TEM) includes vascular, stromal cells, myeloid cells, fibroblasts and extracellular matrix (ECM) proteins [34]. The combination therapy approach to target tumor and also tumor microenvironment is an effective approach for anti-cancer therapy [35]. In addition, combination of different checkpoint inhibitors such as FDA-approved anti-CTLA-4 and anti-PD1 have been proven to be more effective in anti-cancer therapy in clinic [18, 36, 37].
The combination of CAR-T cells with checkpoint inhibitors such as PD-1 or PD-L1 can be used to overcome inhibitory tumor microenvironment by incorporating either PD-1 with CD28 or other co-stimulatory domains or dominant-negative PD-1 receptor with deleted transmembrane and inhibitory intracellular domains [35] or using secreted PD-1 ScFv [38] or silenced PD-1 expression. PD1 dominant-negative receptor engineered into mesothelin CAR structure competed with endogenous PD-1 and saturated PD-L1 expressed on tumor cells preventing inhibitory PD-1/PD-L1 signaling [35].
The “don’t eat me”, CD47/SIRP-1 alpha macrophage immune checkpoint [39, 40, 41, 42, 43, 44, 45, 46, 47, 48] is a very important pathway for regulating survival signaling. The detailed description of each checkpoint signaling regulating recognition of “self” and “not-self” between immune cells and tumor cells is included in the review [18] and in several other reports [34, 49, 50, 51, 52].
The detailed CD80, CD86, CD28, CTLA-4 signaling is discussed in [53, 54, 55, 56, 57]. CD80 (B7-1) is a 33 kDa, 288 amino acid immunoglobulin protein encoded by CD80 gene. CD86 (B7-2) is a 70 kDa, 329 amino-acid immunoglobulin protein encoded by CD86 gene. CD80 and CD86 have been shown to be overexpressed in several types of cancer [54, 58]. The costimulatory ligands CD80 (B7-1) and CD86 (B7-2) are expressed on antigen presenting cells (APC) mediating either costimulatory signaling through CD28 receptor or inhibitory signaling through the cytotoxic T lymphocyte-associated antigen (CTLA-4) (CD152) receptor [59, 60] (Fig. 2). Expression of CD80 and CD86 has been shown to be regulated by epigenetic mechanism such as promoter methylation [54]. Recent report demonstrates that T regulatory cells (or Treg cells) constitutively expressed a cytotoxic T-lymphocyte-associated antigen-4 (CTLA-4) which down-regulated expression of CD80/CD86 co-stimulatory molecules on antigen-presenting cells by CTLA-4-dependent trogocytosis [61]. In addition, decreased CD80 expression by Treg-membranous CTLA-4 blocked cisCD80/PD-L1 heterodimers [61]. The authors suggested that simultaneous inhibition of CTLA-4 and PD-1/PD-L1 can enhance immune responses [61].
A recent study detected that CTLA-4-CD28-CAR-T cells effectively targeted CD80/CD86-positive cancer cells [62]. CTLA4-CAR-T cells effectively killed CD80- or CD86-positive tumor cells (Raji, RL, and NALM6), but not CD80/CD86-negative K562 cells [62]. CTLA-4-CAR-T cells decreased xenograft tumor growth associated with targeting of myeloid-derived suppressor cells (MDSCs) without cytokine release syndrome (CRS) [62]. Thus, authors managed to convert negative CTLA-4-CD80/CD86 signaling to tumor-killing mechanism.
CD70, a member of the tumor necrosis factor superfamily, has been expressed in hematological and solid tumors [63]. CD27 is a receptor for CD70 and provides costimulatory signaling to T cells by inducing cellular proliferation and survival signaling [64, 65, 66, 67]. CD70 has been shown to be overexpressed in mantle cell lymphoma, follicular lymphoma, diffuse large B-cell lymphoma, Hodgkin’s lymphoma, Waldenström macroglobulinemia, and multiple myeloma, but not in normal B cells or T cells [68, 69]. In addition, immunohistochemical staining analysis of CD70 expression in 25 different solid tumor types (N = 496 tissue samples) demonstrated that 43% of solid tumors had highest CD70 expression in renal cell carcinoma (79.5% positivity) and no expression in Kaposi sarcoma [69]. Lymphoma samples demonstrated 58% positivity [69].
In one report CD70-CAR-T cells were generated and demonstrated cytotoxic activity against CD19-negative lymphoma [68]. In other reports CD70-CAR-T cells effectively targeted glioblastoma tumors [70], and also melanoma [71]. Recent report demonstrated effective targeting of CD70 by CD70-CAR-T cells in acute myeloid leukemia (AML) samples [72]. Thus, CD70 can be targeted by CAR-T cells in different hematological and solid tumors.
Intracellular tumor-related antigens can be presented as peptides in the major histocompatibility complex (MHC) on the cell surface interacting with the T cell receptors (TCR) on T cells. Many studies reported targeting MHC-peptide-TCR interactions to block tumor growth using different therapies such as T cell recruiting antibodies, TCR mimicking antibodies, antibody conjugates or autologous genetically-modified effector T cells [73, 74, 75, 76, 77]. The TCR complex contains TCR variable heterodimer (TCR alpha, beta), which binds to the MHC-peptide complex, and a CD3 component which includes gamma, delta, two epsilon and two zeta subunits [78]. Most TCRs tested in clinic were HLA-A2-based and directed against melanoma-associated antigens recognized by T cells such as (MART-1), glycoprotein (gp) 100, carcinoembryonic antigen (CEA), melanoma-associated antigen (MAGE-A3), New York esophageal squamous cell carcinoma antigen (NY-ESO1), or p53 [77]. The TCR mimicking antibodies were generated by either screening of phage display library or through immunizing mice with recombinant MHC-peptide complex which were then used for generating TCR-like CAR-T cells. Several TCR-like CAR-T cells were reported such as MAGE-A1 [79], ESO-1 [80], WT-1 [81], GP100 [82], HMHA1 [83]. CAR-T cells targeting alpha-fetoprotein (AFP)-MHC complex effectively decreased liver cancer growth [84, 85]. AFP-CAR-T cells had high efficacy using Hep G2 tumor xenograft NSG mice model [84]. AFP-CAR-T cells represent an example of CAR-T cells targeting intracellular proteins presented as peptides on the surface of MHC complex. This approach can be applied to other targets.
LAG-3 (CD223) is expressed on T cells, NK cells and Treg cells, binds to the MHC II-peptide complex and blocks function of immune cells [86, 87, 88]. The LAG3 gene is located on chromosome 12 in humans and on chromosome 6 in mice [88]. LAG-3 consists of four extracellular immunoglobulin superfamily-like domains (D1-D4) with additional 30 amino-acid loop in the D1 domain which binds to MHC class II molecules with 100-fold higher affinity than CD4 protein [88]. The extracellular domain of LAG-3 and also cytoplasmic domain have been shown to be involved in the inhibitory signaling of T cells [88, 89].
T cells and primarily NK cells express killer-cell inhibitory receptors (KIR) on their cell surface to bind MHC class I molecules (Fig. 2) [90]. Interaction of KIR with MHC I leads to cell immune surveillance and NK cell-mediated cytotoxicity [90]. Many types of tumors often down-regulate human leukocyte antigen HLA I to escape immune recognition [91]. Recently inhibitory CAR with KIR extracellular domain and intracellular PD-1 domain was introduced into CD19-CAR to decrease 19-CAR-T cell toxicity or “on-target off-tumor” effects [92]. These KIR/PD-1-based CAR-T cells had high efficacy and less toxicity against CD19-positive HLA-C1-negative Burkitt’s lymphoma and did not affect CD19-positive/HLA-C1-positive human B cells [92].
B7H3 (CD276) ligand has been reported to be overexpressed in hematological and solid tumors [93, 94, 95, 96, 97, 98]. B7H3 has been shown to be overexpressed in both tumor and stromal tissues, tumor vasculature, tumor-infiltrating dendritic cells, and macrophages [94]. B7H3 is a 316 amino-acid cell surface protein which is encoded by CD276 gene on chromosome 15 in humans and chromosome 9 in mice [99, 100]. B7H3-CAR-T cells demonstrated significant anti-tumor activity against pediatric solid tumors using Ewing sarcoma, osteosarcoma, or medulloblastoma mouse xenograft models in vivo [101]. In addition, B7H3-CAR-T cells also effectively targeted AML and melanoma tumors in vitro and in vivo using mouse xenograft models [102]. B7H3-CAR-T cells which were administered intracerebroventricularly or intratumorally and demonstrated effective anti-tumor activity against cerebral atypical teratoid/rhabdoid tumors (ATRTs) which are incurable tumors arising in the central nervous system of children under 3 years of age [103]. In addition, B7H3-CAR-T cells were highly effective in a clinical study by inhibiting glioblastoma tumor growth [104]. B7H3-CAR-T cells were also effective against skull chordomas [105]. Another study showed that both B7H3-CAR-T cells and bispecific B7H3-CD3 BITE antibody killed a rare and aggressive subtype of non-Hodgkin lymphoma, extranodal nasal natural killer/T cell lymphoma [106]. The authors compared effects of B7H3-CAR-T cells with B7H3-CD3 BITE antibody and demonstrated very promising results for both treatments.
B7H4 protein belongs to the B7 family proteins and encoded by VTCN1 gene [107]. The hB7H4 sequence encodes a putative protein of 282 amino acids containing several N-glycosylation sites in the extracellular domain [107]. The hB7H4 protein contains a large transmembrane domain and a very short two amino-acid intracellular domain [107]. B7H4 is often overexpressed in many types of solid tumors such as breast, glioma, lung, prostate, melanoma, esophageal, bladder, and particularly in ovarian tumors [108, 109, 110, 111, 112, 113]. B7H4 has very limited expression in normal tissues making it an attractive therapeutic target [109, 114]. B7H4 binding to B7H4 receptor leads to inhibiting T cell proliferation and blocking secretion of cytokines [107, 115]. The B7H4-CAR-T cells inhibited ovarian tumor growth using xenograft mouse model [108]. The authors demonstrate that B7H4-CAR T cells mediated off-tumor toxicity at later time points and suggested that this could be due to expression of B7H4 in healthy mouse tissues [108]. B7H4-CAR-T cells demonstrated high multiorgan infiltration of lymphocyte cells in B7H4-treated mouse model in vivo [108].
PD-L1 (B7-H1), programmed death ligand-1, 40 kDa transmembrane protein [55].
PD-L1 (B7-H1 or CD274) is overexpressed in many types of tumors such as gastric,
lung, breast, ovarian, pancreatic, oral, head and neck, colorectal, brain,
thyroid, liver, kidney, renal, melanoma, skin and hematological cancers [116, 117]. Overexpression of PD-L1 is correlated with worse prognosis in many cancers
[117]. PD-1 (CD279) receptor is expressed on lymphocytes, NK cells, dendritic
cells, B cells [118]. PD-1 receptor has two ligands of B7 family members, PD-L1
and PD-L2 [116]. The anti-PD-L1-CAR-T cells were generated with CAR vector
containing PD-1 extracellular and transmembrane domains, 4-1BB and TLR2
costimulatory and CD3 zeta activation domains and also with CAR vector containing
PD-L1 ScFv with the same costimulatory domains and CD3 zeta cytoplasmic domains
significantly decreased tumor growth of PD-L1-positive solid tumors [119]. Both
dominant-negative PD-1 CAR-T cells and PD-L1-CAR-T cells had synergistic effect
in vivo. In addition, PD-L1-CAR-T cells also lysed PD-L1-positive T
cells in xenograft model [119]. Another report demonstrated that
dominant-negative PD-1 lacking transmembrane and inhibitory cytoplasmic domain
engineered within CAR had high efficacy against solid tumors by blocking
PD-L1/PD-L2 pathways [35]. In another report bispecific PD-L1/c-Met-CAR-T cells
have been shown to effectively inhibit growth of hepatocellular carcinoma [120].
c-Met is a tyrosine kinase Met or hepatocyte growth factor receptor (HGFR) which
is often overexpressed in many types of cancer including hepatocellular carcinoma
[121, 122, 123, 124, 125]. One report showed that c-Met up-regulated transcription of PD-L1
through the MAPK/NF-kappa B pathway promoting the progress of hepatocellular
carcinoma [124]. The bispecific PD-L1/c-Met-CAR-T cells targeting two antigens
PD-L1 and c-Met had specific activity against PD-L1-positive, c-Met-positive
hepatocellular carcinoma xenograft tumors [120]. Another report showed that
PD-L1-CAR-T cells effectively inhibited PD-L1
PD-L1 is overexpressed not only on tumors but also on tumor microenvironment cells such as infiltrating lymphocytes and myeloid cells [14]. The authors used nanobody which consisted of the variable regions of heavy-chain VHH to generate PD-L1-CAR-T cells. Nanobodies are small camelid-derived single domain antibodies which are small and stable with similar to ScFv affinity [14]. VHH doesn’t need folding and linker optimization between VH and VL that can be required for ScFv [14]. VHH is also less immunogenic than mouse ScFv and due to their small size can bind different epitopes [14] The authors demonstrated high efficacy of VHH-based PD-L1-CAR-T cells by targeting PD-L1 on tumor microenvironment using syngeneic melanoma B16 and colon adenocarcinoma MC38 xenograft mice models [14]. Thus, PD-L1 can be targeted with CAR-T cells on both tumors and tumor microenvironment. Generation of the PD-L1-targeted CAR-T cells did not result in fratricide, possibly due to sequestering of the PD-L1 by PD-1 on the T cell surface or due to low level of PD-L1 expression on T cells [14]. The authors were able to use immunocompetent mouse xenograft model with tumor and tumor microenvironment signaling and demonstrate high efficacy of the CAR-T cells.
TIGIT (T cell immunoreceptor with Ig and ITIM domains that can be named as
VSig9, Vstm3, or WUCAM) is a 244 amino-acid checkpoint molecule that belongs to
the poliovirus receptor (PVR)/nectin family [127]. TIGIT binds and competes with
immune-activator CD226 receptor (DNAM-1) for binding to CD112 and CD155 ligands
[128, 129]. The TIGIT mediates immunosuppressive effects by binding to poliovirus
receptor (CD155) and by modulating cytokine secretion by dendritic cells [127].
TIGIT is a promising target for cell therapy as it is overexpressed in multiple
cancer types including colorectal cancer, melanoma, lung adenocarcinoma, breast,
pancreatic, ovarian cancer, and glioblastoma and its expression is correlated
with poor prognosis [130]. Dual PD-1/TIGIT inhibition effectively increased
CD8
T cell immunoglobulin and mucin domain-containing protein 3 (TIM3) encoded by HAVCR2 gene is a member of the TIM family of immunoregulatory proteins (reviewed in [131]). TIM3 has several ligands such as Galectin 9, CEACAM-1 and high mobility group protein B1 [131]. Galectin-9-TIM-3 interaction has been shown to inhibit immune response [131]. TIM3 is expressed on T cells, NK cells and Treg cells [131]. In addition, TIM-3 was shown to be overexpressed in hematopoietic tissues of acute myeloid leukemia (AML) patients [132]. TIM-3 was identified as an AML stem cell surface marker which more highly expressed in leukemia stem cells compared to normal bone marrow hematopoietic stem cells [132]. Recently bispecific CAR-T cells targeting CD13 and TIM3 have been shown to eliminate acute myeloid leukemia cells [133]. The bispecific CAR-T cells (Biss CAR-T cells) targeting CD13 and TIM3 had reduced toxicity compared to single CD13-CAR-T cells [133]. These CAR-T cells targeted tumor cells which expressed both TIM-3 and CD13 proteins but did not kill normal cells that only expressed CD13 [133].
The same approaches can be applied to other immune cell types (macrophages, Treg cells, NK cells, Gamma Delta T cells, dendritic cells, allogenic T/NK cells) with their own pathways [134, 135].
One of the macrophage-related signaling pathways is a CD47-SIRP-alpha pathway. For example, recently CD47-CAR-T cells targeted CD47-positive pancreatic, ovarian and melanoma tumors [17, 136]. Another approach is to engineer CAR-macrophages against solid tumors [137]. The advantage of using macrophages is their effective homing to solid tumors and phagocytotic activity [137].
The same checkpoint pathways can be targeted with CAR-NK cells such as PD-L1/PD-1, KIR, CTLA-4, IL1R8 [138], LAG-3, NKG2A, TIGIT, TIM-3, Siglec 3,9, VISTA and CD161 (reviewed in [139, 140, 141, 142]). All immune checkpoint pathways of NK cells are reviewed in [139]. For example, PD-L1-CAR-NK have been shown to be effective against human and mouse head and neck cancers [143]. The authors demonstrated that PD-L1-CAR NK cells decreased myeloid cells expressing high level of PD-L1 in peripheral blood from patients with head and neck cancer [143].
CAR-gamma delta T cells demonstrated high efficacy against solid and hematological cancers without GHVD (graft vs host disease), and all checkpoint targets can be used for designing CARs similarly as for regular CAR-T cells [144]. Allogenic CAR-T and NK cells without GHVD can be generated using knock-out TRAC, B2M or PD-1 pathways with CRISPR, TALEN or other gene-editing technology [145]. CAR-Treg cells used for therapy are discussed in [146]. There is a cross-talk between T cells, dendritic cells and NK cells that needs to be explored in future studies. Combination therapy approach combining therapies targeting checkpoint pathways has been shown to be very effective to block tumor growth [147, 148, 149].
This report summarizes main checkpoint proteins and CAR-T cells targeting immune checkpoint pathways such as PD-L1/PD-1, B7H3, B7H4, MHC-peptide/LAG3/KIR, TIM-3, CD70 and other shown in Fig. 2. We presented CAR-T cells blocking PD-L1/PD-1, CD112, CD155/TIGIT, B7H3, HLA-peptide, B7H4 and TIM-3 pathways that effectively killed tumors. We presented CAR-T cells which targeted both tumors and tumor microenvironment. Different approaches were discussed such as increasing safety of CAR-T cells using different switches, designing fourth and fifth generation of CAR-T cells by increasing their efficacy by adding cytokine and chemokine secretion, knocking-out checkpoint receptor signaling either with secreted antibodies, ScFv, using dominant-negative checkpoint molecules or silencing RNA. We provided review of other immune cells such as gamma-delta T cells, NK cells, DC and macrophages with similar checkpoint pathways that can be used for designing CARs in these immune cells.
There are several examples of clinical trials that are recruiting patients or active in USA, China and other countries that involve CAR-T cells targeting immune checkpoint pathways (www.clinicaltrials.gov) such as CD19, with PD-1 knock-out engineered CAR-T cells against lymphoma (NCT04213469), mesothelin CAR-T cells with CRISPR-Cas-9 mediated PD-1 and TCR genes knocked-out against relapsed or refractory advanced solid malignancies (NCT03545815). The clinical trial study with B7H3-CAR-T cells recruits patients in Seattle Children’s Hospital, USA with diffuse midline glioma and recurrent or refractory pediatric central nervous system tumors (NCT04185038). Another B7H3-CAR-T cell study recruits patients in Lineberger Cancer Center, Chapel Hill, NC with ovarian cancer (NCT04670068). Several clinical trials using B7H3-CAR-T cells either alone or in combination with temozolomide recruit patients with recurrent glioblastoma in China (NCT05241392), (NCT04077866). Another study recruits patients with CD70-CAR-T cells against hematological cancers in China (NCT04662294).
Future clinical trials will provide more data on CAR-T cells targeting checkpoint pathways and reveal novel biomarkers involved in these pathways. The screening off-tumor on-target effects will be carefully evaluated by different genomics, proteomics, protein arrays and other approaches. The single cell RNAseq report demonstrated importance of genomics approaches to reveal on-target neurotoxicity of CD19-CAR-T cells [150]. The single cell genomic profiling of CAR-T cells before and after treatment will allow to develop biomarkers of response to therapy, resistance players and novel signaling players.
The novel humanized mouse models will be developed to address differences in signaling and targets between murine and human models. The biodistribution of target molecules and biomarkers needs to be carefully examined to more effectively kill solid tumors and provide the best route of CAR-T cell administration (for example, intratumoral, intralymph nodal, intraperitoneal or intravenous). The analysis of mechanisms and signaling driven by tumor microenvironment, involvement of different immune cell types such as NK cells, Treg, macrophages and dendritic cells and checkpoint molecular players will provide basis for more effective tumor killing. Developing biomarkers of response to CAR-T cells targeting checkpoint signaling will require combination of different therapy approaches.
The new ligands for immune receptors on tumor cells are discovered which can be used for CAR-T cells therapy. Novel different CAR-T cells targeting the immune checkpoint ligands on tumors and blocking receptors on immune cells will be developed. The combination of CAR-T cells with checkpoint inhibitors [151, 152], chemotherapy and epigenetics and genetics players will be used to increase efficacy of these therapies.
VG conceptualized and wrote the manuscript.
Not applicable.
We would like to acknowledge all authors of reports whose studies were not included in this review. We would like to thank John Sienkiewicz for professional editing of the manuscript.
This was funded by Promab Biotechnologies.
VG is an employee of Promab Biotechnologies. VG is serving as the guest editor of this journal. We declare that VG had no involvement in the peer review of this article and has no access to information regarding its peer review. Full responsibility for the editorial process for this article was delegated to GP.