
†These authors contributed equally.
Academic Editor: Mateusz Maciejczyk
Background: The mitophagy/NLRP3 inflammasome pathway is a promising therapeutic target for cerebral ischemia-reperfusion (I/R). Panax notoginseng (Burkill) F.H. Chen, one of the most valuable components of traditional Chinese medicine, and Panax notoginseng saponins (PNS), the main active ingredients of P. notoginseng, are patent medicines commonly used to treat cardio- and cerebrovascular diseases. However, their effects on the mitophagy and the NLRP3 inflammasome activation in I/R remain unclear. Therefore, in this study, we investigated how PNS might affect the mitophagy/NLRP3 inflammasome pathway in I/R. Methods: Cerebral I/R injury was induced by middle cerebral-artery occlusion, and expression levels of NLRP3 inflammasome signaling pathway-associated proteins were detected by western blot. We tested I/R injury using a neurological-deficit score, infarct volume, and hematoxylin and eosin staining, after which we detected both mitophagy- and NLRP3 inflammasome-related proteins in PNS-treated rats to determine whether PNS could attenuate I/R injury and the possible mechanisms involved. Results: Our results showed that cerebral I/R could induce activation of the NLRP3 inflammasome, aggravating brain injury, whereas PNS effectively alleviated cerebral I/R injury in rats by inhibiting the NLRP3 inflammasome and promoting mitophagy via the PINK1/Parkin pathway. Moreover, mitophagy inhibited the NLRP3 inflammasome and mediated the anti-injury effects of PNS. Conclusions: In conclusion, PNS could promote mitophagy via the PINK1/Parkin pathway by inhibiting activation of the NLRP3 inflammasome, alleviating cerebral I/R injury in rats.
Stroke is one of the main causes of death and disability in humans, the most common type being ischemic [1]. Currently, the guiding principle for the treatment of ischemic stroke is to restore blood perfusion to ischemic tissue over time [2]. In some cases, recovering tissue blood flow after ischemia cannot effectively restore tissue function but exacerbates its damage, producing an ischemia-reperfusion (I/R) injury [2]. Cerebral I/R injury is the important link in the pathogenesis of ischemic stroke, and its prevention is the key to the current treatment of stroke [2, 3]. Furthermore, cerebral I/R can cause a series of changes at cellular and molecular levels, and the pathogenic mechanisms include mitochondrial-energy metabolism impairment, the toxicity of excitatory amino acids, ion balance disorder, oxidative stress, inflammatory responses, mitophagy, apoptosis, and blood-brain barrier (BBB) destruction [2, 4, 5, 6]. Specifically, the inflammatory response plays an important role in the pathological process of cerebral I/R injury [7].
Inflammasomes are central to the inflammatory response. The nucleotide-binding
oligomerization domain, leucine-rich repeat and pyrin domain-containing 3 (NLRP3)
inflammasome, the most thoroughly studied inflammasome, mediates the incidence
and progression of many neurodegenerative diseases [3, 8]. When stimulated by
intracellular danger signals, NLRP3, apoptosis-associated speck-like protein
containing a Caspase activation and recruitment domain (ASC) and pro-Caspase-1
assemble into a multiprotein complex, termed the NLRP3 inflammasome. This complex
then converts pro-Caspase-1 into Caspase-1, which allows activated Caspase-1 to
cleave pro-interleukin-1
Panax notoginseng (Burkill) F.H.Chen, a highly valued Chinese medicine for treating cardiovascular (CV) and cerebrovascular diseases, is the dried root and rhizome of this species belonging to the Araliaceae family. P. notoginseng is a functional food that is recorded in the ancient book “Compendium of Materia Medica”. Not only does it possess pharmacological activity in tissues including those of the nervous, CV, and immune systems [15], P. notoginseng is also used to make medicinal preparations such as decoctions, tablets, powders, and medicinal liquors [16]. Moreover, Panax notoginseng saponins (PNS) are the main bioactive ingredients of P. notoginseng, which contains diverse monomer saponins; PNS preparations, such as Xuesaitong injection, have been widely used to prevent and treat CV and cerebrovascular diseases [17]. Previous studies, including ours, have shown that PNS has multiple pharmacological activities during ischemic brain injuries, such as anti-inflammatory, antioxidant, and antithrombotic activities, and demonstrates the ability to inhibit apoptosis of neuronal cells and protect the BBB [18, 19, 20]. This evidence suggests that PNS could play a protective role in cerebral I/R through multiple targets. However, further study is needed to confirm whether mitophagy and the NLRP3 inflammasome are involved in the anti-cerebral-I/R injury effects of PNS and whether the mitophagy/NLRP3-inflammasome pathway mediates the mechanism in the protective effects of PNS. Therefore, in the current study, we further investigated whether PNS reduced I/R injury from the perspectives of the NLRP3 inflammasome and mitophagy.
We purchased PNS from Chengdu Manster Biotechnology Co., Ltd. (DST200706-054; Chengdu, China). The major effective constituents of PNS compounds included 6.96% (v/v) Notoginsenoside R1, 34.29% (v/v) Ginsenoside Rg1, 2.65% (v/v) Ginsenoside Re, 41.34% (v/v) Ginsenoside Rb1, and 10.19% (v/v) Ginsenoside Rd [21]. We also purchased an autophagy inhibitor (3-methyladenine [3-MA]; M9281; Sigma-Aldrich, St. Louis, MO, USA), Caspase-1 inhibitor (AC-TYR-VAL-ALA-ASP-C MK [Ac-YVAD-CMK]; SML0429; Sigma-Aldrich), NLRP3 inhibitor (MCC950; S7809; Selleck Chemicals, Houston, TX, USA), mitophagy inhibitor (mitochondrial-division inhibitor 1 [Mdivi-1]; S7162; Selleck), goat anti-rabbit secondary antibody (AP132P; Merck Millipore, Darmstadt, Germany), and goat anti-mouse secondary antibody (AP124P; Merck Millipore, Darmstadt, Germany).
We purchased healthy adult male Sprague Dawley rats weighing 200–220 g (specific-pathogen-free [SPF] grade) from the Experimental Animal Center of HUCM (Animal Certificate No. SYXK, Xiang, 2018-0002). The rats were fed under SPF conditions for 1 week before experiments, with tap water and normal chow supplied ad libitum. The breeding room was provided with natural light and maintained at a temperature of 20–25 °C. All protocols followed the ARRIVE guidelines in terms of study design, sample size, randomization, outcome measures, data analysis, experimental procedures, and reporting of results. This study was approved by the Animal Ethics Committee of Hunan University of Chinese Medicine (HUCM; Changsha, China; Approval No. LLBH-202103290002), in compliance with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH publication no. 85-23, revised 1996). We declare that all methods were performed in accordance with the relevant guidelines and regulations.
We generated an MCAO model as described previously [21]. Rats were anesthetized
with isoflurane (1.5%) in 30%/70% oxygen/nitrous oxide. Their body temperature
was maintained at 37 °C
The length of the line embolism inserted into the artery was 18
We randomly divided the rats into the following treatment subgroups: sham,
model, low-dose PNS (PNS-L), medium-dose PNS (PNS-M), high-dose PNS (PNS-H), PNS
+ 3-MA, PNS + Mdivi-1, Ac-YVAD-CMK, and MCC950 group. The rats were given
microinjections of 3-MA (40 nmol) at a concentration of 5
Following reperfusion for 24 h, we evaluated rats in each group to determine the NDS via the Longa method [21]: 0 = no neurological deficit; 1 = the left forelimb could not be extended when the rat lifted its tail; 2 = circling to the contralateral side when walking; 3 = tilting to the contralateral side when walking; 4 = no spontaneous motor activity, or loss of consciousness.
After reperfusion for 24 h, rats were euthanized by intracardial injection of
500 mg/kg 10% potassium chloride solution; their brains were removed, placed on
ice, and frozen for 15 min at –20 °C. We evenly cut each brain into five
2-mm serial coronal sections. Then, the slices were incubated in 2%
2,3,5-triphenyltetrazolium chloride (TTC) phosphate buffer (BCBP3272V;
Sigma-Aldrich, Saint Louis, MO, USA), immersed in a dark, constant-temperature bath of 37 °C
for 15 min, and stirred gently every 5 min. After staining, we fixed the brain
sections in 4% paraformaldehyde for a minimum of 24 h. The infarct regions
eventually turned white, and the unaffected regions turned red. The brain slices
were then photographed and analyzed using Image-Pro Plus software (Media
Cybernetics, Silver Spring, MD, USA). Additionally, brains were divided into the
ipsilateral hemisphere (ischemic side) and contralateral hemisphere. The
following formula was used to calculate cerebral infarction volume, as described
previously [28]: percentage of infarct volume = (total contralateral hemisphere
volume-non-infarcted ipsilateral hemisphere volume)/(total contralateral
hemisphere volume)
Following reperfusion for 24 h, rats (n = 8) were anesthetized and euthanized
via intracardial injection of 500 mg/kg 10% potassium chloride solution and then
killed using rapid decapitation. We then fixed the brains in 4% paraformaldehyde
and sliced them into sections with 3-mm thickness, cutting coronally through the
optic cross-plane. The slices were dehydrated using gradient alcohol, made
transparent with xylene, embedded in paraffin, and finally cut into
3-
We used WB to detect NLRP3 inflammasome levels and mitophagy-related proteins.
After reperfusion at 24 h, in accordance with the literature [29], we weighed and
homogenized the cerebral cortex of the ischemic penumbra and then added it to 10
volumes of pyrolysis liquid (P0013; Shanghai Biyuntian Biotechnology Co., Ltd.,
Shanghai, China), a protease inhibitor cocktail (B14002; Bimake, Houston, TX,
USA), and phosphatase inhibitors (B15002; Bimake, Houston, TX, USA). The lysate
was then centrifuged at 12,000
We analyzed the collected data using IBM SPSS version 22.0 (IBM Corp., Armonk,
NY, USA). The results are expressed as mean
We detected NLRP3 inflammasome–related protein expression using WB. The results
showed that proteins related to NLRP3 inflammasome were significantly elevated in
the model group, as compared with the sham group. Expression of NLRP3, Caspase-1,
pro-IL-1
 Fig. 1.
Fig. 1.NLRP3 inflammasome was activated, and suppressing it reduced
injury during cerebral I/R in rats. (a) WB bands of NLRP3 inflammasome-related
proteins in different groups. (b–i) Comparisons of relative protein expression
levels among groups (n = 5). (j) TTC-stained brain
slices; pale areas are infarcted (n = 8). * p
To determine the role of NLRP3 inflammasome activation in cerebral I/R, MCAO rats received i.p. injections of Ac-YVAD-CMK and MCC950, after which we ascertained their infarction volumes and NDS. The results showed that infarction volumes were larger (Fig. 1j,k), and NDS was higher (Fig. 1l), in the model group than in the sham group. Conversely, infarction volumes and NDS were significantly decreased in both Ac-YVAD-CMK and MCC950 groups versus the model group (Fig. 1j,k,l). These findings showed that cerebral I/R could activate both the NLRP3 inflammasome and Caspase-1 and that inhibiting these could reduce cerebral I/R injury in rats.
To reveal the protective effects of PNS on I/R injury, we recorded the infarction size, NDS, and cell damage rate after PNS treatment. The results of TTC staining showed that the infarct volume was larger in the model group than in the sham group whereas the administration of PNS reduced infarct volume in MCAO rats in a dose-dependent manner (Fig. 2a,b). Neurological function in the sham group was normal (NDS = 0) whereas the model group displayed clear neurological deficits and had a noticeably higher NDS than the sham group (Fig. 2c). Additionally, the NDS and rate of cell damage in the PNS groups were significantly reduced compared with the model group (Fig. 2c,d). From H&E staining, we also found a reduced degree of cell damage that was directly proportional to PNS dose (Fig. 2d). Thus, the conclusion can be drawn that PNS could effectively reduce cerebral I/R injury in rats.
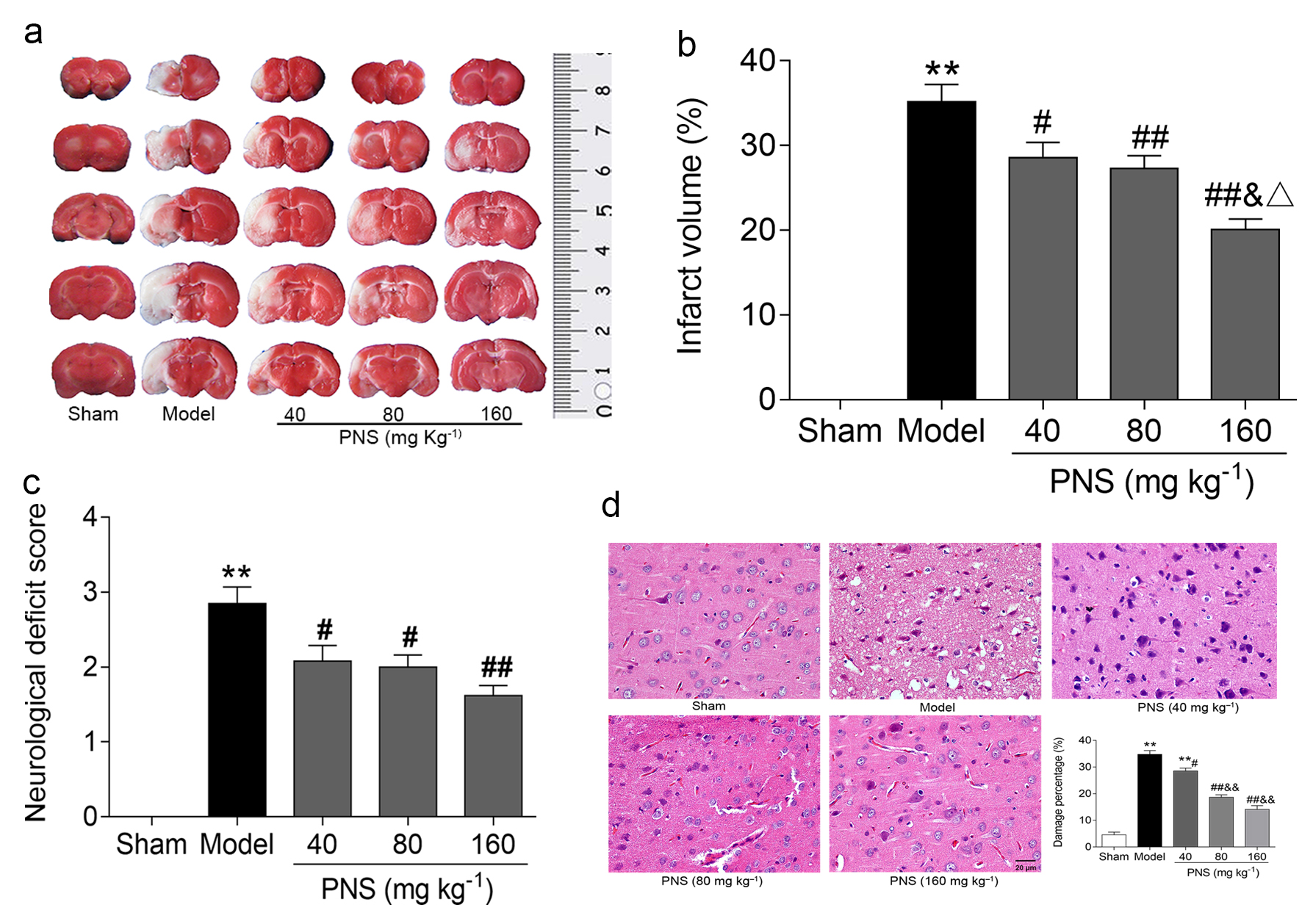 Fig. 2.
Fig. 2.PNS reduced cerebral I/R damage in rats. (a) TTC-stained brain
slices from the sham, model, and PNS (PNS-L, -M, and -H) groups; pale areas are
areas of infarction (n = 8). (b) Statistical chart of determined cerebral-infarct
volumes in each group (n = 8), created using Image-Pro Plus software. (c)
Statistical bar chart of evaluated NDS in each group (n = 13). (d) Pathological
changes in the cerebral cortex and statistical map of the cell injury rate in
each group (n = 8). * p
We used WB to assess changes in the NLRP3 inflammasome and its downstream
proteins in rats treated with or without PNS to explore the function of PNS on
NLRP3 inflammasome activation. Compared with the sham group, the expressions of
NLRP3, ASC, pro-Caspase, Caspase, pro-IL-1
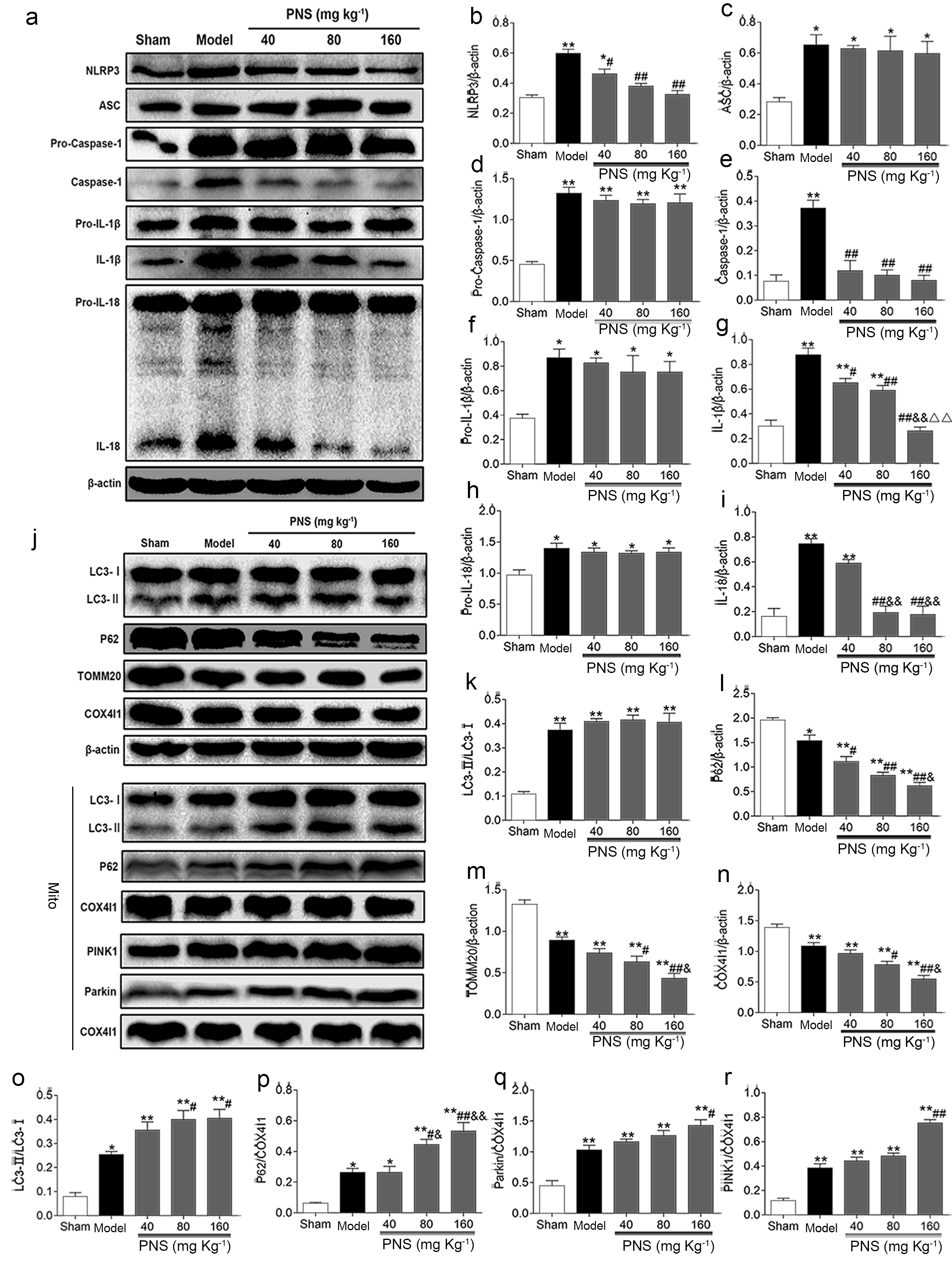 Fig. 3.
Fig. 3.PNS inhibited NLRP3 inflammasome activation and activated
mitophagy via the PINK1/Parkin pathway in rats. (a,j) WB analysis of relative
proteins in the sham, model, and PNS (PNS-L, -M, and -H) groups (n = 5). (b–i,
k–r). Densitometry scanning of band densities was used to quantify expression of
proteins in each group using Quantity One software
(n = 5). * p
To discern the role of mitochondrial autophagy in PNS-mediated neuroprotection, we evaluated changes in protein markers of mitochondrial autophagy via WB. The results indicated that mitochondrial p62 protein, as well as LC3-II/LC3-I ratio of both total-brain and mitochondrial proteins, increased significantly in MCAO rat brain tissues; expression of total-brain P62 and mitochondrial COX4I1 and TOMM20 exhibited the opposite changes (Fig. 3j–p). Furthermore, there was a significant reduction in cerebral P62 level, with insignificant changes in the ratio of cerebral LC3-II/LC3-I after PNS administration. Moreover, in the PNS-M and PNS-H groups, expression levels of COX4I1 and TOMM20 proteins were significantly decreased compared with the vehicle group, and the LC3-II/LC3-I ratio and p62 protein level in mitochondria were also increased (Fig. 3j–p). Additionally, with decreasing expression of total-brain P62, levels of mitochondrial P62, COX4I1, and TOMM20 were concurrently increased in the PNS-H group compared with the PNS-L group (Fig. 3j,m,n,p).
We then examined the effect of PNS on PINK1 and Parkin expression levels. The WB results showed significantly increased mitochondrial PINK1 and Parkin in both the model and PNS-H groups compared with the sham group (Fig. 3j,q,r). These results showed that PNS inhibited activation of NLRP3 inflammasome and activated PINK1/Parkin–mediated mitophagy during cerebral I/R.
We administered autophagy inhibitor 3-MA or the mitochondrial-autophagy
inhibitor Mdivi-1 to rats just after MCAO to confirm the effect of PNS-mediated
mitophagy on NLRP3 inflammasome activation. As shown in Fig. 4, expression levels
of cerebral NLRP3, Caspase-1, IL-1
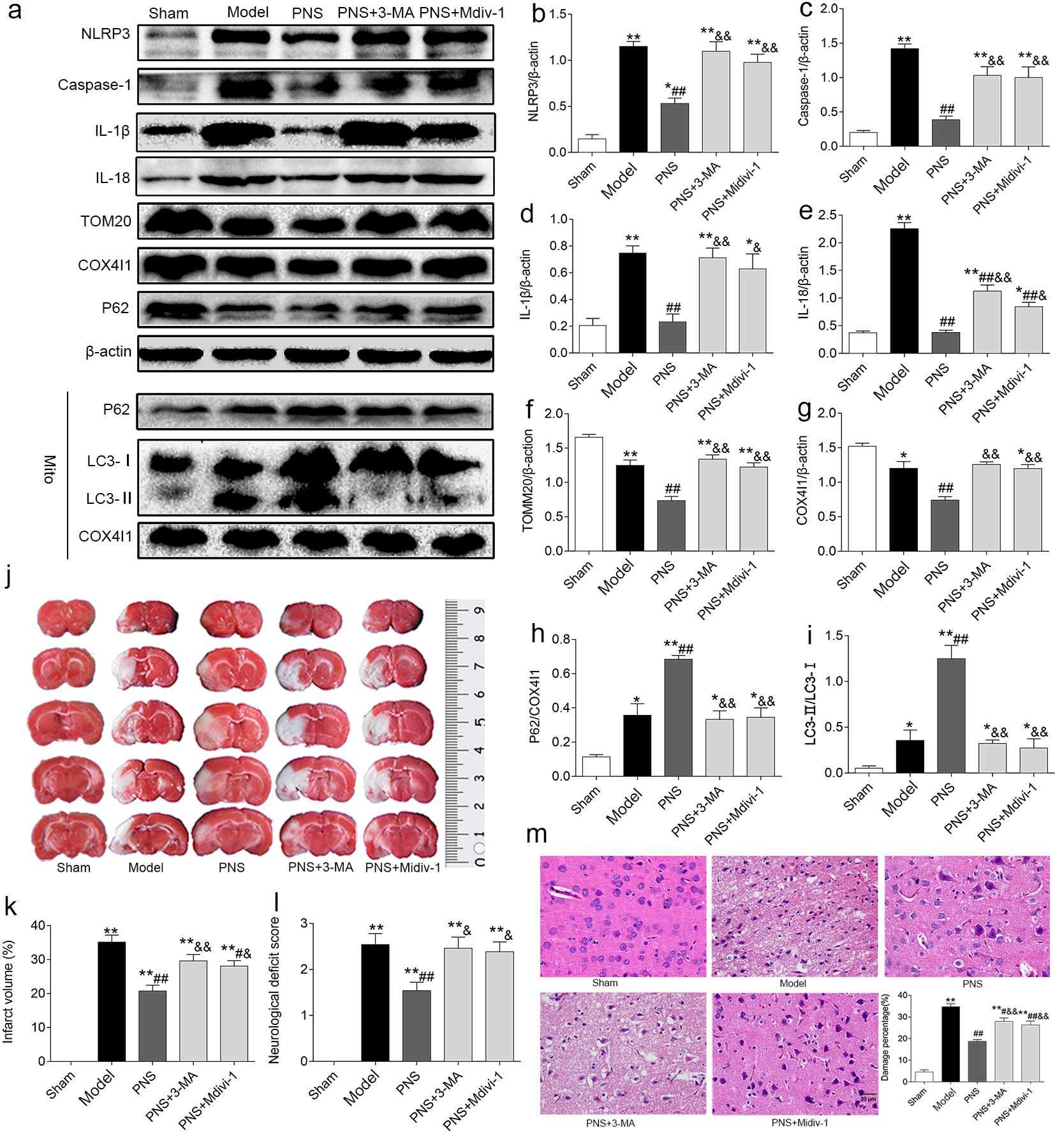 Fig. 4.
Fig. 4.PNS-mediated mitophagy suppressed NLRP3 inflammasome activation
and alleviated cerebral I/R injury in rats. (a) WB analysis of relative protein
levels in the sham, model, PNS-M, PNS + 3-MA, and PNS + Mdivi-1 groups (n = 5).
(b–i) Densitometry scanning of band densities was used to quantify the
expression of proteins in each group (n = 5). (j)
TTC-stained brain slices from the sham, model, PNS-M, PNS + 3-MA, and PNS +
Mdivi-1 groups; pale areas are areas of infarction. (k) Statistical bar chart of
determined cerebral-infarct volumes in each group (n = 8), created using
Image-Pro Plus software. (l) Statistical bar chart of NDS evaluated according to
the Longa method in each group (n = 13). (m) Pathological changes in the cerebral
cortex and statistical map of the cell injury rate in each group (n = 8).
* p
To further identify the involvement of mitophagy in the protective effect of PNS, we detected cerebral I/R injury after PNS co-administration with 3-MA or Mdivi-1 in each group. The results showed that cerebral-infarct volume and NDS were significantly elevated in the vehicle group compared with the sham group (Fig. 4j–l). In addition, these two indicators were significantly decreased by medium-dose PNS but increased again after the subsequent addition of 3-MA or Mdivi-1 (Fig. 4j–l). Finally, we also observed similar results in cell damage rates via H&E staining (Fig. 4m). These results suggested that mitophagy mediated the inhibitory effect of PNS on activation of the NLRP3 inflammasome and had anti-brain injury effects in rats with cerebral I/R.
We demonstrated that NLRP3-related protein expression reached its highest levels after 12 or 24 h of reperfusion, which showed that cerebral I/R induced both priming and activation of the NLRP3 inflammasome. Additionally, our results showed that inhibition of the NLRP3 inflammasome by Ac-YVAD-CMK and MCC950 ameliorated cerebral I/R injury in rats. Moreover, these results were consistent with findings from previous studies [30, 31, 32, 33, 34]. We found that the activation of the NLRP3 inflammasome and Caspase-1 aggravated cerebral I/R injury in rats after MCAO and its inhibition could alleviate cerebral I/R injury. Several studies have revealed the promotion of mitophagy-suppressed activation of the NLRP3 inflammasome in cerebral I/R [10, 11, 12, 13].
PNS exert protective effects in cerebral ischemia through multifactorial pathways [10, 20, 35], which has also been confirmed in animal experiments, in vitro cell experiments, and clinical data [18, 35]. However, the mechanism that PNS regulates NLRP3 inflammasome activation via mitophagy in cerebral I/R needs further research. This was therefore our focus in the current study.
We first demonstrated that PNS attenuated cerebral I/R injury in MCAO rats by
decreasing the cerebral-infarct volume, NDS, and cell damage rate. Additionally,
our results showed the activation of NLRP3 inflammasome and a significant
decrease of Caspase-1, IL-1
Evidence produced by other researchers has provided insight into the idea that mitophagy can be activated and plays a protective role in cerebral ischemia-reperfusion injury [7, 8]. Inhibiting mitophagy can reduce cerebral ischemia-reperfusion injury [36], but such differences may be attributed to varying times of ischemia, reperfusion, or cell types. However, in the present constructed brains of male SD rats, 2-h arterial occlusion and 24-h reperfusion promoted mitophagy and inhibited the activation of inflammasome, which attenuated cerebral ischemia-reperfusion injury. Therefore, we clarified whether PNS regulates mitophagy during cerebral I/R. Our further investigation revealed that in terms of total proteins after PNS treatment, LC3-II protein levels demonstrated no significant change whereas P62 protein levels decreased. However, levels of COX4I1 and TOMM20, as well as those of mitochondrial LC3-II and P62, increased significantly. This suggested that PNS could selectively promote mitophagy rather than autophagy and that the relevant mechanism might be related to PNS promoting P62 translocation to mitochondria. This speculation aligned with the findings of a previous report [37]. Furthermore, it has been reported that PNS activates mitophagy to protect the kidneys in cisplatin nephrotoxicity [38]. Multiple signaling pathways are involved in regulating mitophagy, including Parkin-dependent and Parkin-independent pathways, such as BNIP3/NIX and FUNDC1. In these regulatory pathways, Pink1/Parkin has been most thoroughly studied. After mitochondrial damage, the ability to degrade PINK1 is weakened. PINK1 accumulates on the mitochondrial outer membrane, phosphorylates Parkin, and recruits Parkin from cytoplasm to mitochondria. The activated Parkin is activated by the signal adaptor protein p62, which recognizes and binds SQSTM1. p62 can recruit ubiquitinated substances to autophagosomes by binding to LC3, eventually leading to mitochondrial degradation by lysosomes [39]. After cerebral ischemia-reperfusion, fluorescence observation of the penumbral area of the rat cortex reveals PINK1 accumulation on the mitochondrial outer membrane, Parkin translocated to mitochondria, and elevated autophagy-related proteins [40], consistent with our experimental results. Moreover, we demonstrated that PNS could further elevate PINK1 and Parkin protein levels in the cerebral mitochondria of MCAO rats, meaning that mitophagy activated by PNS was promoted through the PINK1/Parkin pathway.
In addition, when PNS were administered, NLRP3, Caspase-1, IL-1
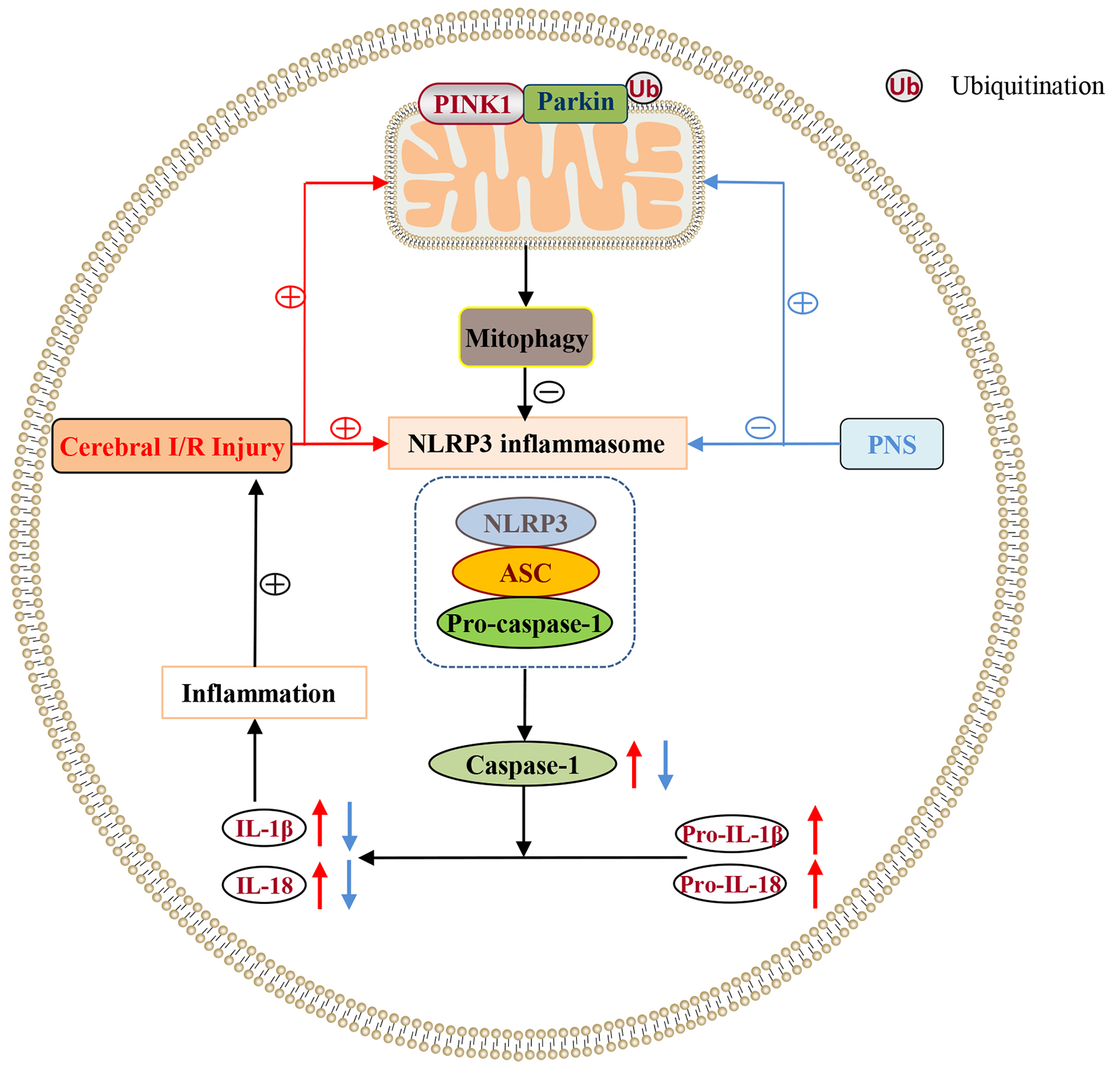 Fig. 5.
Fig. 5.Mechanism of PNS against cerebral I/R injury mediated by the mitophagy/NLRP3 inflammasome pathway. PINK1 accumulated in the outer membrane of mitochondria and recruited Parkin from the cytoplasm to transfer it to the outer membranes of mitochondria. Parkin ubiquitinated mitochondrial-surface substances and mediated mitophagy, which further inhibited activation of the NLRP3 inflammasome. The first part of this experiment demonstrated that mitophagy was activated via the PINK1/Parkin pathway during cerebral I/R, which could promote priming and activation of the NLRP3 inflammasome, aggravating inflammation and, therefore, injury. In the second part of the experiment, PNS was shown to activate mitophagy via the PINK1/Parkin pathway, thus inhibiting NLRP3 inflammasome activation and alleviating cerebral I/R injury. However, the mechanism of how PNS promotes mitophagy and what role is played by the PINK1/Parkin pathway in this mechanism needs further clarification, which are limitations of this study. Further studies will be carried out on other pathways, such as ferroptosis. Revealing the anti-CIRI mechanism of PNS based on the interaction between various pathways will be an important direction of future research.
This study extended previous observations that NLRP3 inflammasome activation exists in cerebral I/R and aggravates cerebral I/R injury. We concluded that PNS could promote mitophagy via the PINK1/Parkin pathway, inhibit NLRP3 inflammasome activation, and reduce cerebral I/R injury.
BT and QX designed the research study; QX, ZK, CL and BT performed the research; CL analyzed the data; QX, ZK and CL wrote the manuscript. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript.
Ethical approval for the study was obtained from the Hunan University of Chinese Medicine (HUCM) Animal Ethics Committee (Changsha, China; Approval Number: LL2021042803).
Not applicable.
This work was supported by the following: National Natural Science Foundation of China (grant. no. 81503385); Hunan Provincial Natural Science Foundation of China (grant. no. 2021JJ30505); Hunan Provincial Health and Family Planning Commission Project (grant. no. B202303079716), Scientific Research Foundation of the Hunan University of Chinese Medicine [grant. no. 202024].
The authors declare no conflict of interest.
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.