
Epithelial-to-mesenchymal transition (EMT) is a fundamental cellular phenomenon that plays an intrinsic role in development, tissue repair, and cancer progression. EMT is tightly regulated by transcription factors that alter gene expression to promote epithelial to mesenchymal phenotype. EMT is also regulated by a diverse array of cytokines and growth factors whose activities are deregulated during malignancy. EMT enables tumor cells to exist in various intermediate states along the epithelial-mesenchymal phenotypic axis that transit from cancer stem cells (CSCs) to circulating tumor cells (CTCs). Recent studies have revealed the importance of CSCs in tumor promotion, invasion and metastasis. The relapsed tumors encompass CSCs which are resistant to radiotherapy and chemotherapy. In this review, we have summarized our current understanding of the molecular mechanisms that regulate EMT induced CSC phenotype. We have highlighted studies implicating the function of TGF-β, Wnt, and Notch regulated non-coding RNAs in driving EMT promoting CSC self-renewal. Finally, we discuss how the EMT and CSCs cause drug resistance with the hope to overcome such resistance as a possible approach for cancer treatment.
Epithelial-mesenchymal transition is a cellular phenomenon that allows a stationary polarized epithelial cell to undergo various morphological changes to assume a migratory, invasive mesenchymal cell phenotype. The initial observation that epithelial cells lose their epithelial characteristics and adopt mesenchymal characteristics was made by Dr. Elizabeth Hay in 1980, who observed epithelial to mesenchymal phenotypic changes in the primitive streak of chick embryos (1). The process now known as epithelial to mesenchymal transition (EMT) is an intrinsic cellular phenomenon reminiscent of embryonic development and tissue repair, which occurs also during diverse disease processes including cancer invasion, organ fibrosis (2). EMT is orchestrated by a group of EMT transcription factors (EMT-TFs) belonging to SNAIL, TWIST and ZEB families. The activation of EMT-TFs occurs in response to various signaling pathways such as Transforming Growth Factor β (TGF-β), Wnt and Notch. These extracellular signals activate the intracellular kinase cascades which ultimately induce the expression of EMT-TFs. EMT and mesenchymal to epithelial transition (MET) which involves the conversion of mesenchymal cells to epithelial cells are consistent with the idea of cell plasticity. Cancer metastasis is driven by EMT mainly by conversion of cancer cells to CSCs, which are capable of traversing tissue boundaries, entering into the blood vessels, localizing and growing in other tissues to form metastatic foci. Therefore, collectively targeting CSCs and cells with activated EMT signaling may provide possible treatment options for cancers that become resistance to the available treatment strategies. In this review, we particularly focus on EMT, plasticity, CTCs, cancer stemness with an emphasis on the the role of cellular signaling in development of EMT in head and neck squamous cell carcinoma (HNSCC) and breast cancers (BC).
Epithelial and mesenchymal cells vary from each other in many aspects. Epithelial cells form an organized layer of cells that are closely adjoined by specialized membrane structures such as tight junctions, gap junctions, communication junctions, adherens junctions, and desmosomes. Epithelial cells exhibit an apical-basal polarity, while mesenchymal cells do not form an organized cell layer and exhibit a front-back end polarity. EMT involves a series of events during which epithelial cells lose their epithelial characteristics and adopt mesenchymal characteristics (2). The core changes that occur during EMT are the reduction in E-cadherin (CDH1) expression that disrupts cell-cell adhesion (3), loss of apico-basolateral polarity, reorganization of the cytoskeleton resulting in the formation of migratory mesenchymal cells with invasive properties (Figure 1). The downregulation of CDH1 is accompanied by reorganization of desmosomal proteins (plakoglobin, desmogleins, and desmoplakins), tight junction proteins and cell polarity proteins. Consequently, there is an increase in expression of mesenchymal markers such as neural cadherin (N-cadherin), vimentin, integrins, fibronectin, and matrix metalloproteinases (MMPs) allowing cells to detach from each other and penetrate the basement membrane (4-6). Hitherto, it was believed that the EMT transition occurs completely. However, EMT is not a complete process, where cells exhibit both epithelial and mesenchymal characteristics along a gradient of epithelial to mesenchymal transition (7).
 Figure 1
Figure 1Tumor progression with increase in EMTTFs. Deregulation in the cell cycle check points and signaling pathways that regulate proliferation/differentiation in the cells of the normal epithelium leads to tumor initiation. These initiated cells proliferate uncontrollably and then undergo EMT transition where they lose their epithelial phenotype and attain mesenchymal phenotype with increase in EMT-TFs such as SNAIL, SLUG, TWIST1/2 and ZEB1/2. (EMT: Epithelial to mesenchymal transition; TF’s: Transcription factors).
EMTs can be classified into three different biological subtypes based on the biological context in which they occur (8, 9). Type-1 EMT occurs during embryogenesis that gives rise to the mesoderm, endoderm, and mobile neural crest cells. Type-2 EMT takes place during wound healing, tissue regeneration, and organ fibrosiswhereas Type-3 EMT is associated with cancer progression which is a key event in the cancer invasion and metastasis.
The role of EMT and its reverse process, mesenchymal to epithelial transition (MET), has been extensively studied in a variety of tissue remodeling events, such as mesoderm formation, neural crest development, heart valve development, secondary palate formation etc. Mesoderm formation and neural crest development occur during early embryonic development, whereas heart valve development and secondary palate formation occur in relatively well-differentiated epithelial cells, which later become defined mesenchymal cell types. The primary mesenchyme is formed from the upper epiblast epithelium (1). The second wave of EMT occurs during the formation of neural crest cells, which further migrate to form the peripheral nervous system (10). Furthermore, the dorsal part of the somite undergoes EMT to give rise to muscle and connective tissue of dermal layers of the skin (12, 13). EMT-like changes also occur during palatal formation that separates the nasal and oral cavities. In aduts, EMT regulate placental formation and production of fibroblast during inflammation and wound healing (14,15,16). During wound healing, non-motile epithelial cells undergo EMT that enable them to migrate to a wound site followed by proliferation. Further, these mesenchymal cells revert to the epithelial state via MET involving a process known as re- epithelialization (17).
EMT-TFs tightly regulate the complex process of EMT that comrprisesof zinc-finger proteins such as SNAILs (SNAI1, SNAI2/SLUG, SNAI3/SMUC), the E-box-binding proteins ZEBs (ZEB1/TCF8 and ZEB2/SIP1), the basic helix-loop-helix protein TWISTs (TWIST1 and TWIST2), and the fork-head box proteins FOXCs (FOXC1and FOXC2). EMT-TFs in co-oepration with other TFs regulate the expression of target genes either by functioning as repressors of epithelial genes or inducers of mesenchymal genes (18, 19). For instance, SNAIL induces expression of other EMT-TFs such as SLUG, ZEB1, and TWIST1. Moreover, SNAIL, SLUG and ZEB1 regulate the expression of another EMT -TF TCF3 (20, 21). On the other hand, extracellular signals such as TGF-β, epidermal growth factor (EGF), fibroblast growth factor (FGF), hepatocyte growth factor (HGF), Wnt and ECM components activate, one or more of these EMT-TFs to induce EMT events (22). EMT-TFs are further regulated by non-coding microRNAs (miRNAs). The best-characterized miRNA regulating EMT are the members of miR-200 family, which downregulate ZEB1 and ZEB2. Further, miR-34 family attenuates the expression of SNAIL. The expression of miR-200 and miR-34 are in turn repressed by ZEB1/ZEB2 and SNAIL respectively implying the presence of double-negative feedback loops between EMT and miRNA (23-26). EMT-TFs such as SNAIL, SLUG, TWIST, ZEB1, and ZEB2 also suppress the expression of E-cadherin, which is considered as a hallmark in EMT (4).
EMT-TFs can be divided into two groups based on the mechanism by which they downregulate the expression of E-cadherin. The first group comprises SNAIL, SLUG, ZEB1, ZEB2, E47, KLF8, and Brachyury that directly interact with the E-cadherin gene promoter to suppress its expression. For instance, SNAI1 repress the transcription of E-cadherin by binding to the E-box region on the E-cadherin promoters followed by recruiting histone modifiers such as HDAC1 & HDAC2 (mSIN3A-histone deacetylase 1 and 2), PRC2 (polycomb repressive complex 2) and LSD1 (lysine-specific demethylase1).The second group comprises TWIST1, FOXC2, Goosecoid, E2-2, SIX1 and PrRRX1 that induce EMT without directly binding to the E-cadherin gene promoter. TWIST1 upregulates the PRC1 component BMI1 and acts cooperatively with BMI1 to repress E-cadherin expression (22, 27-29).
EMT-TFs also down-regulate claudin, occludin, desmoplakin, and plakophilin proteins, which are essential for the maintenance of tight junctions or desmosomes in epithelial cells (4). Conversely, EMT-TFs activate mesenchyme-associated genes such as N-cadherin, fibronectin, and vimentin. SNAIL and ZEB2 also activate matrix metalloproteinases (MMPs) expression facilitating the degradation of the basement membrane and promoting cell invasion. Recent studies have revealed the importance of other TFs such as HMGA2, Goosecoid, KLF8, FOXC2, SIX1, Brachyury, PIT-1, E47, E2-2, ZEPPO, GATA3, and PRRX1 during EMT (30).
Lappin and colleagues stated that “The poetic rhythm of the developmental program does not appear to resonate with the same harmony in the adult form” and EMT in pathological conditions is not an exception. Pathological EMT involves the same genetic and molecular players involved during development; however, they lack the intricate coordination. Induction of EMT in epithelial tumor cells results in disruption of intercellular junctions, such as E-cadherin, claudins, occludins, desmosomes and acquisition of mesenchymal markers such as α-SMA, FSP1, vimentin, and desmin (31). These cells eventually enter into subsequent steps of the invasion-metastasis cascade, i.e., intravasation, transport through the circulation, extravasation, formation of micro-metastases ultimately leading to colonization (Figure 2) (32).
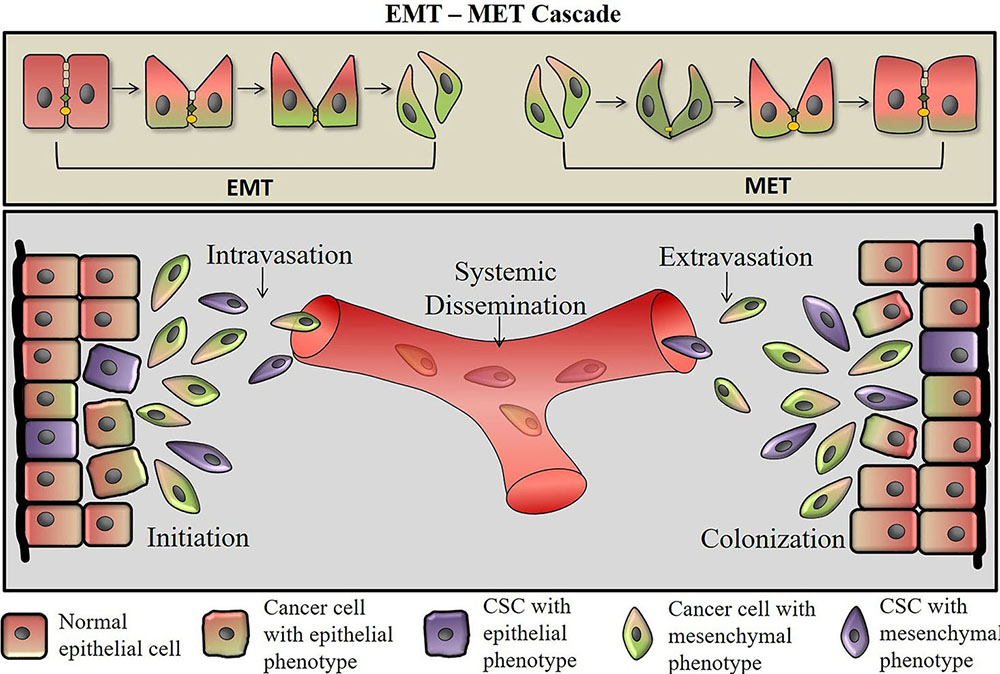 Figure 2
Figure 2EMT and MET in cancer metastasis. Within the CSCs, induction of EMT leads to the expression of EMT- inducing transcription factors (EMT- TFs) ZEB, SNAIL, SLUG, and TWIST, which inhibit the expression of genes associated with the epithelial state and concomitantly activate the expression of genes associated with the mesenchymal state. During EMT, the cancer cells become motile and acquire invasive capacities. They secrete matrix metalloproteases that help in intravasation. The cells then undergo systemic dissemination and reach secondary tumor sites where they undergo mesenchymal-epithelial transition (MET). EMT is a reversible process and mesenchymal cells can revert to the epithelial state by undergoing MET. These cancer cells then colonize and form tumors similar to that of parent tumors at the secondary tumor site. EMT: Epithelial to mesenchymal transition; TF’s: Transcription factors; CTC: Circulating tumor cells; CSCs: cancer stem cells; MET: mesenchymal-to-epithelial transition.
During intravasation, tumor cells detach from the tumor site,invade the walls of the blood vessel and enter into the blood stream, which are termed as circulating tumor cells (CTCs). Recent evidence suggests that the presence of CTCs with mesenchymal characteristics is associated with tumor dissemination and metastasis (34). Further, detection of CTCs in epithelial cancers such as breast cancer and HNSSC is considered as a blood-based biomarker for poor prognosis. CTCs are capable of re-seeding at the primary site suggesting their role in local tumor recurrence (33). Moreover, during extravasation, metastasizing CTCs shed their mesenchymal phenotype via a MET in the course of secondary tumor formation.
Initiation of tumor growth at the secondary site is the rate-limiting step in metastasis. Therefore, a key feature of a successful metastatic cell is cellular plasticity i.e., the ability to undergo EMT and subsequently transit to MET in the appropriate microenvironments.
EMT was initially thought to be a process in which epithelial cells transform completely to mesenchymal cells with a loss of E-cadherin (epithelial marker) expression andagain of N-cadherin expression (mesenchymal marker). However, recent studies have revealed that during EMT, cancer cells simply do not switch from epithelial-to-mesenchymal state but rather they exist in a plethora of intermediate cellular phenotypes, which depend on the regulation of specific genetic programs and the tumor microenvironment (35). An intermediate state between epithelial and complete mesenchymal states is known as partial/hybrid EMT state. During partial EMT, cancer cells acquire mesench ymal traits that enable them to depart from the primary tumor site and survive in the circulation. Subsequently, these cells revert to the epithelial state during metastatic colonization and development of secondary tumors. For instance, the sub-population of CTCs in breast cancer patients showed bi-phenotypic E+/M+ state with clear epithelial histology, both in the primary tumors and draining lymph nodes (36). Further, single-cell transcriptomics in HNSCC showed partial EMT as an independent predictor for the following such as nodal metastasis, grade, and adverse pathologic features. Cancer cells exhibiting partial EMT were spatially localized at the leading edge of the tumor (37).
Tumor cells with hybrid epithelial and mesenchymal phenotypes were more efficient in reaching the circulation, colonizing the lungs and forming metastases (38). A hybrid EMT phenotype observed in ovarian cancer was associated with increased cancer cell stemness, whereas complete epithelial or complete mesenchymal phenotype was associated with both decreased stemness and tumorigenicity (39). Moreover, co-expression of epithelial and mesenchymal markers in other human cancers including colorectal, lung, and pancreatic cancers have been associated with poor survival with resistance to therapy (39).
Cancer cells that exhibit a degree of phenotypic plasticity and undergo the reverse process of the EMT i.e. MET), are more efficient at seeding metastases (Figure 2). Complete loss or forced overexpression of EMTTFs, such as ZEB1, TWIST and SNAIL leads to complete epithelial or mesenchymal phenotype, which is unfavorable for metastasis and CSC maintenance. Constitutive activation of TWIST1in human mammary epithelial cells (HMECs) transformed cells to a migratory and non-proliferative state whereas its transient activation enabled long-term invasive growth due to the coexistence of epithelial and mesenchymal traits (28).
Several reports showed higher levels of EMT markers in the tumor cells not only promote tumor invasion and metastasis to a distant site but also contribute to tumor relapse by virtue of their relative resistance to chemotherapy and radiation (5). These cells acquire stem-like characteristics, which serve as critical drivers of tumor progression that are termed as cancer stem cells (CSCs) (40). CSCs lie at the apex of tumor cell hierarchy capable of self-renewal and giving rise to various differentiated cell types (41). Tumor initiation and relapse are strongly linked with the biology of CSCs (40). Here, we focus on the link between EMT and cancer stemness.
Adult stem cells (SCs) have the remarkable capacity to self-renew and differentiate into various cell lineages to either maintain normal tissue homeostasis or repair the injured tissue SCs give rise to transit-amplifying (TA) cell populations, which terminally differentiate after several rounds of divisions, which are eventually lost from the tissue. (42). Dick and colleagues reported cancer cells expressing stem cell markers (CD34+/CD38-/Lin-) , which showed self-renewal property similar to normal stem cells, which are termed as leukemic SCs (LSCs).LSCs express similar genes as those expressed by Hematopoietic Stem Cells (HSCs), suggesting that LSCs are regulated by similar molecular mechanisms as those of normal stem cells (43-45).Similarly, in solid tumors, it was first demonstrated in breast cancer that only a fraction of cancer cells (CD44+/CD24−/low) have a higher ability to generate tumors upon transplantation into immune-deficient mice (46). In addition, cancer cells expressing aldehyde dehydrogenase (ALDH1) possess tumor-initiating capacity (47). Tumor cells simultaneously expressing both CSC markers (i.e., CD44+/CD24−/low and ALDH1+) displayed higher tumorigenic potential as compared to cells expressing either CD44+CD24−/low only or ALDH1+ only (47). Subsequently, CD44, CD24, and ALDH were used to isolate CSCs from a wide variety of cancers, including those of the HNSSC (48), pancreas (49), colon (50), lung (51), ovary (52) and prostate gland.
A certain degree of plasticity exists within the progenitors or differentiated cells that enable them to form an SC-like state. For instance, upon ablation of airway epithelial stem cells, fully committed secretary cells de-differentiated into functional epithelial stem cells (53). Similarly, ablation of Lgr5 positive intestinal SCs using mice expressing diphtheria toxin receptors in Lgr5 cells resulted in the dedifferentiation of progenitors into an SC-like state, making them capable of sustaining intestinal homeostasis. This suggests that there exists an alternative cellular mechanism by which tissues can regenerate even after stem cell loss (54). This plasticity of normal progenitor/committed cells appears to be reflected in cancer cells where non-CSCs can convert into CSCs under certain conditions. For instance in melanoma, histone H3 lysine-4 (H3K4) demethylase, JARID1B positive population are considered to be CSCs due to their slow-cycling nature and high tumorigenic potential. Nevertheless, Jarid1b-negative melanoma cell can be tumorigenic and re-express Jarid1b upon transplantation implying the conversion of non-CSCs into CSCs (55). Furthermore, induction of a core set of transcription factors (TFs) such as POU3F2, SOX2, SALL2, and OLIG2 in fully differentiated glioblastoma cells (56) and OCT3/4, SOX2 and KLF4 in human colon cancer cells (57) was sufficient to reprogram these differentiated cells into CSCs. Additionally, the plasticity of non-CSCs of human basal breast cancers enabled them to switch from a non-CSC to CSC state. The observed cell plasticity was dependent on ZEB1, whose promoter is maintained in a bivalent chromatin configuration. Upon TGF-β stimulation, the ZEB1 promoter converted from a bivalent to active chromatin configuration. As a result, ZEB1 transcription increased, fueling the conversion of non-CSCs to CSCs (58). Therefore, the plasticity of CSCs and non-CSCs should be an important determinant while developing treatment modalities.
A striking similarity between the gene-expression profiles of cells undergoing EMT and stem cells suggests a close connection between EMT and stemness of CSCs. The link between EMT and cancer stemness was first shown in the differentiated mammary epithelial cells that gave rise to CD44high/CD24low cells upon TGF-β treatment or forced expression of E-cadherin transcriptional repressor, SNAI1 (59). EMT-associated factors such as TWIST1/2, FOXC2, SNAI1, ZEB2, VIM and FN1 were expressed at higher levels in CD44+/CD24−/low as compared to more differentiated epithelial CD44-/CD24+ cells. Furthermore, breast cancer cells disseminated in the circulation and bone marrow were enriched for the CD44+/CD24− antigen subset. EMT-TF’s such as SNAIL, SLUG and TWIST etc. were responsible for the generation and maintenance of CSCs in many other tumor types, including colorectal (60), hepatocellular carcinoma (61), ovarian (62), etc. In HNSCC, the expression of EMT-TFs was increased in CSCs thereby suggesting EMT-TFs as the common factor for metastasis and stemness of cancer cells. Moreover, many studies found a positive correlation between stem-cell like phenotype and metastatic potential of cancer cells. CD271, a stem-cell marker, was associated with higher SNAI2/SLUG expression. Moreover, the higher invasive and metastatic phenotype was also observed in CD271-expressing cell lines, patient-derived xenografts and primary human HNSCC (63). In a study examining different grades of human oral Squamous Cell Carcinoma (OSCC) samples showed higher CD44 that was concomitant with elevated expression of Vimentin and VE-cadherin in higher grade carcinoma indicating a functional relationship between cancer stem cells and metastasis (64).
In an early-stage OSCC, tumors with higher expression of stem cell markers such as CD133, NANOG, and NOTCH-1 showed an invasive phenotype with metastatic potential (65). Furthermore, CD44+/CD45-/CD31- cells isolated from primary OSCC showed a marked downregulation in cell adhesion genes such as PCDH18, MGP, SPARCL1 and KRTDAP as compared to non-stem cells (66). These studies point to the fact that CSCs exhibit a mesenchymal phenotype and a greater metastatic potential. The downregulation of MMPs in OSCC cell lines led to a subsequent reduction in stem-cell markers such as ABCG2 and CD44, indicating that inhibition of metastasis-aggravating molecules impacts the stemness of tumor cells (67). AF4/FMR2 family member 4 (AFF4), a core component of SEC (Super elongation complex), increased ALDH1 and also enhanced the migration and invasion capability of HNSCC cell lines. Moreover, immune-histochemical analysis of OSCC tissue samples concluded that stem-cell markers ALDH1 and ABCG2 were higher in the aggressive and metastatic tumor (68).
miRNAs play a crucial role in regulating EMT induced cancer stemness. The evolutionary conserved miR-200 is the most widely studied miR in the regulation of EMT and stemness. It has been associated with stem-like cell signatures by regulating the expression of BMI1, NOTCH1 and LIN28B expression (69-71). miR-200c inhibited the clonal expansion of breast cancer cells and suppressed the growth of embryonal carcinoma cells in vitro. In addition, it inhibited the ability of normal mammary stem cells to form mammary ducts as well as tumor formation by human BCSCs in vivo. Three miR clusters, miR-200c-141, miR-200b-200a-429, and miR-183-96-182 were also down regulated in human mammary stem cells and human BCSCs and embryonal carcinoma cells (70). During EMT induction, OSCC cells switch from CD44 variant (CD44v) to CD44 standard form (CD44s) by suppressing miR-220c, which in turn increases the availability of ZEB1 to repress E-Cadherin expression (72).
These studies bolster the fact that stemness and metastatic properties of a tumor are indeed intricately linked and CSCs emerge as the major player contributing to the process of EMT. Moreover, induction of EMT in tumor cells not only promotes stemness properties, tumor cell invasion and metastasis but also contributes to drug resistance. Therefore, an increase in our mechanistic understanding of the EMT–CSC link will allow the development of newer therapies for eradication of tumors to improve the overall survival of patients that have been diagnosed with cancers. Thus, targeting the EMT to eliminate CSCs offers a promising avenue for the improvement of cancer therapy.
EMT is fundamental to both embryogenesis and tumor metastasis. EMT is activated by major signaling pathways such as Wnt, TGF-β, and Notch, etc. The binding of ligands to their associated receptors expressed by normal and neoplastic epithelial cells triggers a cascade of intracellular signaling events that culminate in activating the EMT program. Here, we discuss some of the major signaling events that are mainly involved in EMT of HNSCC and breast cancer.
Wnt-β-catenin signaling pathway is an evolutionarily conserved pathway involved in early embryonic development, self-renewal, tissue homeostasis and tumorigenesis. Wnt signaling is mediated by Wnt ligands (19 Wnt ligands) and their associated Wnt frizzled receptors (10 Fzd receptors) and Wnt inhibitors (families of Sfrps, Dkks and Wif1) (Figure 3) (73, 74). During embryonic development, Wnt3 is essential for the formation of the primitive streak, which is required for the formation of an anterior-posterior neural pattern (75). Additionally, Wnt-11 and Frizzled-7 (Non-canonical Wnt signaling) are crucial for the migration of neural crest cells (76). For instance, Sfrp1, a natural Wnt antagonist, is down regulated in various human cancers due to promoter hypermethylation such as breast cancer, oral squamous cell carcinoma (OSCC), lung cancer and hepatocellular carcinoma (HCC), etc (145,146,147,148) Recently, our laboratory showed that CSCs of Sfrp1-/- murine skin SCC showed an upregulation of EMT markers such as Twist1, Twist2, Snail, Vimentin, Zeb1 and Sox2, stem cell marker. Further, these data was validated in human HNSCC and breast cancer tumor samples, which showed decreased Sfrp1 while increased Sox2 expression thereby establishing an inverse correlation, with overall poor survival (149). Further, Wnt signaling is required for the proliferation and migration of epidermal stem cells during the wound repair process (77, 78).
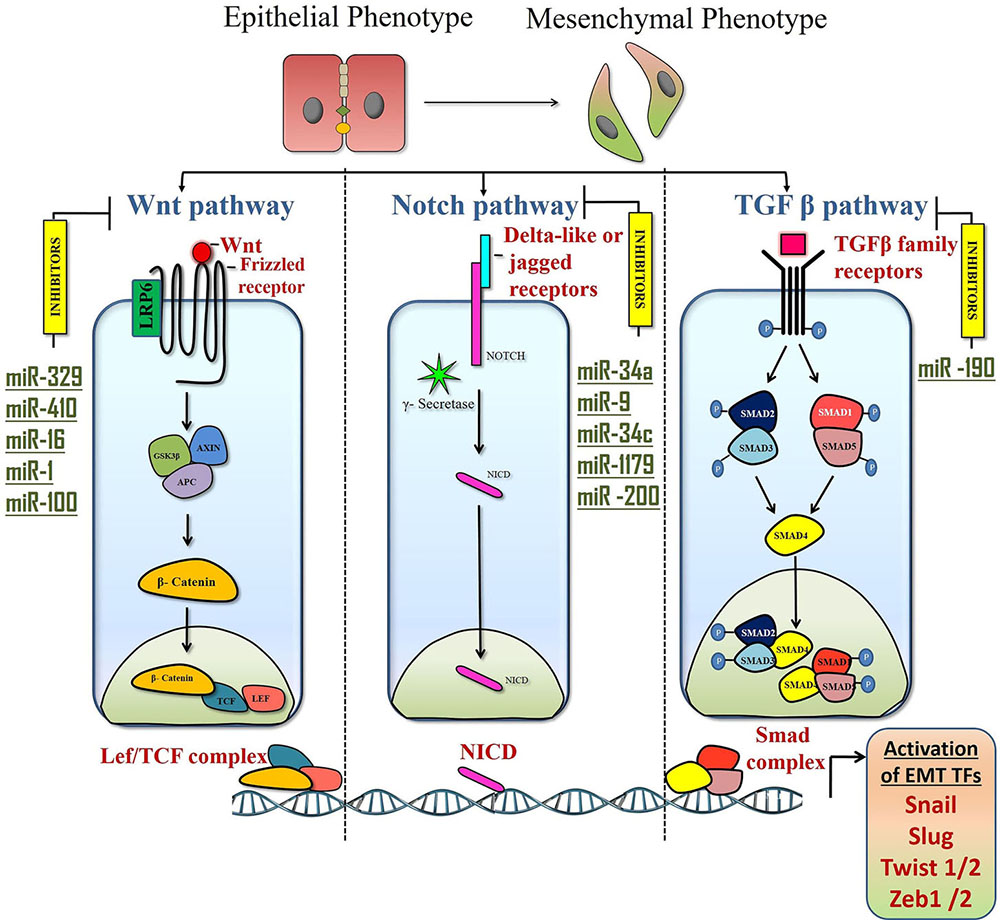 Figure 3
Figure 3Signaling pathways that activate EMT and their regulation by miRs. Multiple cell-intrinsic signaling pathways cooperate to induce the expression of EMT TFs such as ZEB, SNAIL, SLUG and TWIST that induce the transition to the epithelial cell state to mesenchymal cell state. Canonical WNT pathway is activated upon binding of canonical WNT ligands to the Frizzled family of membrane receptors. This leads to the release of β- catenin from the destruction complex. β- Catenin then translocate to the nucleus, where it binds to the transcription factors TCF (T cell factor) and LEF (lymphoid enhancer- binding factor) to activate genes that drive EMT. Micro RNAs such as miR-329, miR-16 and miR-410 down regulate WNT signaling thereby regulating EMT in HNSCC whereas miR-1 and miR-100 target Wnt signaling to inhibit EMT in breast cancer. The Notch pathway is activated upon binding of the Delta-like or Jagged family of ligands to the Notch receptor. This binding activates γ-secretase which cleaves Notch to form NICD, which enters the nucleus to function as a transcriptional co-activator.miR34a, miR-1179, miR-200, miR- 9 and miR-34c are some of the miRs that target Notch to inhibit breast cancer invasiveness. Binding of the TGF-β proteins to the TGF-β family of receptors leads to receptor phosphorylation and activation of SMAD 2/3 or 1/5 which forms complexes with Smad4 and enters into the nucleus and then activates the EMT program. miR190 targets TGF-β signaling and inhibits breast cancer metastasis. EMT: Epithelial to mesenchymal transition; TF’s: Transcription factors; CTC: Circulating tumor cells; CSCs: cancer stem cells; miRs: microRNA; HNSCC: Head and neck squamous cell carcinoma.
In HNSCC, Wnt ligands and its regulators were reported to enhance tumor cell migration and invasion. In HNSCC, Wnt-5b overexpression enhanced invasion and migration of tumor cells by regulating MMP10 expression through MEF2A (79, 80). Further, a decrease in inhibitory Wnt-7a and an increase in Wnt-5a/fzd5 expression resulted in overall poor survival of HNSCC patients (81). Additionally, Wnt signaling also inhibited anoikis and increased cell invasion in HNSCC. Conversely, higher expression of Dickkopf protein-3 (DKK3) in HNSCC patients showed a high metastatic rate and overall poor survival (82). MicroRNAs also play a pivotal role in regulating WNT signaling either activating or inhibiting EMT. miR-21 downregulates DKK2, Wnt antagonist, there-by activating invasion in OSCC cells (83). Conversely, higher expression of miR329 and miR410 down-regulated WNT-7B that is involved in activating proliferation and invasion of OSCC (84). Additionally, overexpression of miR-16 inhibited OSCC growth by inhibiting Wnt-β-catenin signaling (85).
During breast cancer metastasis, canonical Wnt signaling helps the cancer cells to stay in an epithelial/mesenchymal (E/M) hybrid state by differential expression of EMT-inducing transcription factors (86). Wnt-Axin2-Gsk3β cascade regulates SNAI1 activity there-by regulating EMT in breast cancer (87). Further, Wnt-3 over-expression confers EMT like properties and trastuzumab resistance to Her-2 overexpressing breast cancer cells (88). Moreover, miR-374a targets negative regulators of Wnt signaling such as WIF1, PTEN, and WNT5A, thereby activating Wnt signaling and distant metastasis in breast cancer patients (89). Furthermore, active miR-1 down-regulated breast CSC stemness, proliferation, and migration by inhibiting the Wnt/β-catenin signaling through regulation of FZD-7 and Tankyrase-2 (TNKS2) (90). miR-100 suppressed migration and invasion of MDA-MB-231 cells by targeting WNT8 and inhibiting Wnt β-catenin pathway (91). Additionally, long non-coding RNAs, lncRNA-UCA1 enhanced EMT in MDA- MB-231 cells by targeting GSK-3β and activating Wnt pathway (92).
TGF-β signaling plays an instrumental role during early development as well as during cancer metastasis. TGF-β signaling is mediated by type I and type II receptor serine/threonine kinases. Once the ligand binds to the type II receptor, it trans-phosphorylates the type I receptor, which then phosphorylates cytoplasmic Smad2/Smad3 or Smad1/Smad5 based on the type of ligand. Subsequently, activated Smad2/3 or Smad1/5 form complexes with Smad4 and then enter into the nucleus, which binds to chromatin and regulates expression of genes that play critical roles in the control of cell proliferation, differentiation (including EMT), apoptosis and cell migration (Figure 3) (93).
During the early development of vertebrate embryos, neural crest EMT is regulated by dose-dependent actions of bone morphogenetic proteins (BMP), members of the TGF-β superfamily, and a cohort of transcription factors, including paired-box, high-mobility group (HMG), winged-helix transcription factors and Snail (94). Further, mice lacking the TGF-β3 gene exhibit a cleft palate suggesting that TGF-β3/ LEF1 cascade plays a major role in epithelial cell transformation during palatal EMT (95).
In tumors, TGF-β signaling leads to the activation of EMT through several different mechanisms. A high level of TGF-β1 results in an increased expression of SNAIL through SMAD2, 3 and 4, which enhanced the expression of MMP-9 and MMP-2, followed by an increased invasiveness in OSCC cells (96), (97). Alternatively, TGF-β1 can also regulate the expression of SNAIL independent of SMADs through ERK1/2 , which then can activate EMT in HNSCC (80). Further, TGF-β1 also regulates Slug through ERK1/2, which enhances invasion in OSCC cell lines through an increased expression of MMP-9 (98). Moreover, co-stimulation of OSCC cells with TGF-β1/EGF results in EMT phenotype with up-regulation of vimentin and down regulation of E-cadherin (99). Hypoxia also plays an important role in inducing EMT. Overexpression of hypoxia/HIF-1α enhances Slug1 expression resulting in decreased E-cadherin and enhanced invasiveness in HNSCC patients (100). Apart from TGF-β ligands, microRNAs play a significant role in regulating cancer metastasis. Overexpression of miR-96-5p leads to increased cell migration and radio/chemo-resistance in HNSCC cells by targeting PTEN and activating the PI3K-AKTsignaling pathway (101).Furthermore, low expression levels of miR-153, a negative regulator of SNAI1 and ZEB2, increases metastasis and poor prognosis in oral cancer patients (102). Also, overexpression of miR-200c and miR-141 significantly increased the E-cadherin expression by targeting ZEB1 and ZEB2 thereby reducing the migration capacity of HNSCC cells (103), (104). The overexpression of Long non-coding RNA (lncRNA) such as EPB41L4A-AS2 showed decreased invasion and migration in OSCC cells (105).
In breast cancer, TGF-β1 promoted CCR7 expression in cancer cells and CCL21 expression in lymphatic endothelial cells, resulting in chemotactic migration of cancer cells toward lymphatic endothelial cells (106). Further, TGF-β1 also regulates EMT by regulating several micro RNAs. miR10b overexpression induced by TGF-β1/twist1 positively regulates breast cancer cell migration and invasion (107). TGF-β signaling mediated expression of Slug binds to the miR-203 promoter and inhibits its expression thereby enhancing the metastatic properties of breast cancer cells (108). In addition, TGF-β signaling inhibits miR-584 expression and enhances cell migration in breast cancer cells (109). In certain cases, chemotherapy can also enhance CSC like phenotype and invasion. For instance, Paclitaxel treatment activates TGF-β signaling, which increases cancer stem-like cells that are drug-resistant and more invasive (110). Conversely, miRNAs also negatively regulate TGF-β signaling and inhibit EMT. In the case of breast cancer, miR190 inhibits metastasis by targeting SMAD2 and inhibiting TGF-β signaling (111). Further, miR-200 inhibits EMT and cancer cell migration by directly targeting E-cadherin transcriptional repressors such as ZEB1 and ZEB2 in mammary tumors (112). Additionally, OVOL2 (Ovo-like zinc finger protein) expression inhibits TGF-β signaling at multiple levels: inhibition of Smad4 mRNA expression blocking the binding between Smad4 and target DNA, and interfering with complex formation between SMAD4 and SMAD2/3, thereby inhibiting EMT during mammary tumor metastasis (113).
The Notch signaling is a highly conserved pathway, which is primarily involved in controlling cell fate decisions, differentiation and proliferation (114), (115). It is mediated by jagged family and delta-like ligands binding to their receptors (NOTCH1–NOTCH4).This binding triggers a series of proteolytic cleavage events by γ-secretase that lead to the generation of the active Notch intracellular domain (NICD), which then translocate to the nucleus leading to expression of target genes by associating with other transcriptional activators (Figure 3) (114). There is a surplus of evidence suggesting the involvement of Notch signaling in embryonic development and cancer metastasis. In embryonic development, Notch is involved in endocardial maturation and EMT initiation through Snail for proper cardiac development (116).
In OSCC cell lines, Notch signaling induces EMT by activation of Snail in a hypoxic environment (117). All the members of Notch family such as NOTCH1, 2 and 4 are involved in tumor progression and metastasis except for Notch 3, which induces tumor senescence in HNSCC. Notch1 regulates invasion and metastasis in HNSCC by activating C-MYC, which induces Snail thereby increasing the expression of Vimentin and decreasing E-cadherin expression (118). NOTCH2 expression was higher in LSCC (laryngeal squamous cell carcinoma) cells that undergo lymph node metastasis as compared to node negative LSCC (119). NOTCH4 and HEY1 expression were associated with an EMT phenotype as well as increased invasion and cell migration in HNSCC (120). Further cross-talk between TGF-β and Notch signaling also activates EMT in HNSCC. For instance, TGF- β activates the transcription factor ZEB1 to repress NOTCH3, thereby limiting terminal differentiation and drives NOTCH1-mediated EMT to generate tumor-initiating cells characterized by high CD44 expression in OSCC (121).
In breast cancer, Notch signaling activates EMT through more than one mechanism. NOTCH1 signaling activates SLUG expression that promotes migration, and invasion of breast cancer cells (122). Further, the overexpression of NOTCH1 activates STAT3 that showed induction of CSC like phenotype and EMT in breast cancer cell lines (123). Hypoxia also plays an important role in activating Notch signaling. Hypoxia induced activation of Notch signaling enhances the expression of SNAI1 and lysyl oxidase (LOX),which further activate EMT in breast cancer cell lines (124). Inhibition of NOTCH3 significantly reduced the self-renewal and invasive capacity of TNBC cells and ERα+ cells. Conversely, NOTCH3 suppressed EMT and breast cancer metastasis by activating GATA binding protein 3 (GATA-3) transcription (125). Several miRNAs were reported to have a negative impact on the Notch-induced EMT. miR34a overexpression led to notch-1 inhibition and activation of p53 and caspase3/9 in MCF-7 cells (126). Overexpression of miR-1179 inhibited the expression of NOTCH 1, NOTCH 4 and HES1 that significantly suppressed breast cancer cells proliferation, migration, and invasion (127). Further, miR-200 family miRNAs suppress the Notch signaling by targeting Notch pathway components, such as JAG1 and the mastermind-like Notch co-activators, MAML2 and MAML3 (128). Overexpression of Notch signaling can also be suppressed by the induction of miR-9 and miR-34c expression, thus reducing metastatic behaviors in TNBC (129).The Mastermind-Like Protein 1 enhanced E-cadherin expression and decreased the rate of cell migration in breast cancer cell lines by inhibiting Notch signaling (130).
The success of current anti-cancer therapies is hindered due to therapy resistance. Such acquired resistance is partly driven by a subpopulation of cells residing within the tumor called as cancer stem cells. CSCs are more resistant to drugs and toxins due to the high expression of several ATP-binding cassette (ABC) transporters, increased DNA repair capacity, deregulated signaling, and resistance to apoptosis. Recent studies have shown that the EMT program is a critical regulator of the CSC phenotype. Patients manifesting both the CSC and EMT phenotypes are have low disease free survival (DFS) as they are unresponsive to standard chemotherapies. Signaling networks regulated by microRNAs and EMT-TFs linking the EMT and cancer stemness can serve as possible targets. Currently, many clinical trials are underway that are testing the efficacy of Wnt, Notch and TGF-β inhibitors. Small molecules such as Galunisertib, TEW-7197 and PF-03446962 are inhibitors of TGF-β receptor type 1. ETC-1922159, an inhibitor of the membrane-bound O-acyltransferase (MBOAT) porcupine (PORCN) in the endoplasmic reticulum, results in the inhibition of post-translational palmitoylation of Wnt ligands and their secretion. OMP-54F28, a recombinant fusion protein that binds to Wnt ligands and blocks Wnt signaling. The major treatment modalities in the treatment of HNSCC, include surgery, radiotherapy, neoadjuvant chemotherapy or a combination of two or more of these modalities (131). However, the 5-year survival rate of this disease has not changed evidently from the past few decades (132). Hence, a detailed understanding of the tumor initiation, cancer stem cell regulation and their metastasis is required for better cancer treatment.
The most commonly used chemotherapeutic drugs in HNSCC include platinum-based cytotoxic drugs like cisplatin and carboplatin, which binds to DNA and inhibit DNA replication. Taxanes such as Docetaxel,that inhibits mitotic spindle assembly (133) and methotrexate (134), which inhibits dihydrofolate reductase (DHFR) thereby inhibiting DNA and RNA synthesis (135). Further targeted therapies include monoclonal antibodies such as cetuximab, which inhibits binding of EGF to its receptor, by specifically binding to EGFR (136). The major drawback for cetuximab is that the pre-existing IgE antibodies against cetuximab cause hypersensitivity in patients. In addition, mAb's (monoclonal antibodies) such as nivolumab and pembrolizumab are involved in inhibition of binding of PD1 to its ligands PDL1 and PDL2, thereby augmenting immune responses by T-cell proliferation and cytokine production (134, 137). However, through all these treatment procedures the overall survival of the patient has not improved significantly. Therefore, new treatment modalities are to be developed to treat HNSCC. The role of signaling pathways in CSCs and their regulation by miRNAs has been well studied in the past decade. Higher expression of miR329, miR410 and miR-16 showed inhibition of Wnt signaling thereby inhibiting proliferation and invasion of OSCC (84). Further, overexpression of miR-200c and miR-141 significantly reduced the migration capacity of HNSCC cells by targeting ZEB1 and ZEB2 activated by TGF-β signaling (103, 104). Hence, developing new treatment modalities using miRNAs for the treatment of HNSCC might give us an edge to treat HNSCC.
Breast cancers are commonly treated with cell cycle inhibitors such as abemaciclib (inhibits cyclin-dependent kinases CDK4 and CDK6), paclitaxel (interferes with microtubular function, chromosomal segregation) and cepecitabine (inhibits the synthesis of thymidine monophosphate required for DNA synthesis). Monoclonal antibodies such as pertuzumab and trastuzumab bind to Her2 and inhibit Her2-Her3 dimerization and block downstream PI3K/AKT signaling pathway. Despite the different treatment modalities, drug resistance is common in all breast cancer types. For instance, mucin 4 and CD44/hyaluronan polymer complex was shown to mask the Her2 epitope, thereby making breast cancer cells resistant to trastuzumab (138). Since triple-negative breast cancer (TNBC) cells do not express ERα+ or PR or show HER2 amplification, there are no targeted therapies for this breast cancer subtype (138). One of the major problems in using taxanes (paclitaxel, doxitaxel) as chemotherapeutic option is the drug efflux. This is mediated by adenosine triphosphate (ATP)-binding cassette proteins that utilize ATP hydrolysis to efflux a variety of drugs making breast cancer resistant to drugs, irrespective of breast cancer subtypes (139).
Non-coding miRNAs regulate signaling pathways involved in cancer stemness, drug resistance, and EMT. miR-1 and miR-100 down-regulate breast CSC stemness, proliferation and migration by inhibiting the WNT/β-catenin signaling (90, 91). miR190 and miR-200 inhibits breast cancer metastasis by targeting SMAD2 and ZEB1 and ZEB2 respectively thereby inhibiting TGF-β signaling (111, 112). An overexpression of miR34a, miR-9, miR-34c, and miR-1179 leads to Notch signaling inhibition and suppression of breast cancer cell proliferation, migration and invasion (127), (126), (129). Harnessing these microRNAs to develop treatment modalities would provide better outcomes in treating breast cancer.
Advancements in basic and translational cancer biology have enabled us to understand the mode of actions and the outcomes of cancer therapies. However, we are not yet successful in coming up with fruitful strategies to treat cancer. The conventional therapies targeting the bulk tumor cells do not take into account the impact of tumor heterogeneity, mainly caused by CSCs, on the efficacy of chemotherapeutic drugs. CSCs generated by EMT induction acts as a source for cancer to recur due to their highly drug-resistant nature. Several strategies can be adopted for targeting EMT induced cancer stemness such as, specifically targeting cells that exhibit CSC phenotype and have undergone EMT, reversing the process of EMT by differentiation induced therapies or targeting the signaling processes that induce EMT.
Recent studies have identified CD44, ADLH1, ABCG2 and many others as CSC markers in HNSCC, but a definitive marker is still lacking (140). There can be differences in stem cell population between different individuals based on co-expression of different markers; hence, patient-specific CSC identification in a specific cancer subtype can help in better disease prognosis and patient outcome. For instance, combination therapies of Cetuximab along with platinum-based drugs yielded positive results, but tumor recurrence occurred in many (141). Hence, a better understanding of tumor heterogeneity and cancer stem-cell subset identification in different patients receiving treatment will help in prognosis and in developing patient-specific therapy based on HNSCC’s CSC subset identification. Similarly, in breast cancer, positive ALDH1 staining was found to be a prognostic marker for both TNBC and non-TNBC subtypes. ALDH1 was also observed to be significantly correlated with resistance to chemotherapy in TNBC patients along with cleaved caspase-3 and cyclooxygenase-2 (142). More recently, MALDI-TOF analysis of breast cancer tissues from three different regions in TNBC patients revealed higher expression of nine novel genes significantly correlated with poor recurrence-free survival (RFS) and a higher hazard ratio in TNBC patients as compared to estrogen-receptor-positive tumors. These studies indicate the fact that an intensive search for a better and definite prognostic marker differentiating between cancer subtypes could help us in designing better treatment strategies and targeted therapies.
During the last decade, a significant amount of research is focused on miRNA mediated regulation of EMT-TFs and cancer stemness. miRNA, Let 7c, a known tumor suppressor, whose expression was reduced in OSCC CSCs. Further, Let 7c also increased the anticancer functions of tamoxifen and reduced the number of \CSCs sensitizing cells to therapy-induced repression in an estrogen receptor (ER) dependent manner. Notably, Let 7 decreased the tumor forming ability of estrogen-treated breast CSCs in vivo and suppressed Wnt signaling. This further consolidated the previous hypothesis that Let 7 decreases the self-renewal ability, contributing to reduced tumor formation ability of stem cells (143). 5-Aminolevulinic acid-mediated photodynamic therapy (ALA-PDT) serves as an adjuvant therapy against HNSSC via eliminating the CSCs property. The anti-CSCs effect of ALA-PDT is due to an elevation of miR-145 (144). These evidence suggest that miRNAs play a significant role in regulating signaling and EMT- induced stemness in HNSCC and breast cancers. Hence, re-expression of specific miRNAs that are lost in these cells will help in eliminating the stem-like cells that are the "root cause" of tumor development, maintenance, recurrence, and metastasis. Therefore, we propose that the development of inhibitors of EMT or CSC-targeted therapies holds a promising future for cancer treatment.
Manan Y Parmar, Saima Shaikh have contributed equally to this work. SR is supported by UGC, India and RRS is supported by ACTREC and DBT, India for their doctoral fellowships.
ABC
ATP Binding Cassette
Atp-Binding Cassette Sub-Family G Member
Aldehyde Dehydrogenase-1
Breast Cancer Stem Cells
Bone Morphogenic Protein
Cyclin Dependent Kinase
Cancer Stem Cells
Circulating Tumor Cells
E-Cadherin
Di-Hydrofolate Reductase
Dickkopf Protein 3
Extracellular Matrix
Epidermal Growth Factor
Epithelial To Mesenchymal Transition
Emt-Transcription Factors
Fibroblast Growth Factor
Frizzled Receptor
Histone Deacetylaces
Hepatocyte Growth Factor
High Motility Group
Head & Neck Squamous Cell Carcinoma
Histine-3 Lysine-4
Leucine rich repeat containing G protein-coupled Receptor 5
Long non-coding RNA
Membrane-Bound O-Acyl-Transferase
Mesenchymal To Epithelial Transition
Matrix Metalloproteinase
microRNA
Notch Intra-Cellular Domain
Polycomb Repressive Complex-2
Transforming Growth Factor-?
Triple Negative Breast Cancer