
 , Xiaodong Tian 1,*
, Xiaodong Tian 1,*1 Department of General Surgery, Peking University First Hospital, 100034 Beijing, China
Academic Editor: Yingqun Wang
Abstract
Pancreatic cancer is still one of the most perilous malignant tumors with a very poor prognosis. Despite the progress in the diagnosis and treatment of pancreatic cancer, the overall 5-year survival rate after diagnosis is less than 10%. The pathogenesis of pancreatic cancer has not been fully clarified, but multiple factors are involved. The poor efficacy of traditional therapies for pancreatic cancer is mainly related to complex tumor microenvironment. In recent years, accumulating studies have demonstrated the role of autophagy and apoptosis triggered by endoplasmic reticulum stress in pancreatic cancer. In particular, unfolded protein response is activated by endoplasmic reticulum stress and plays an important role in the modulation of complex pancreatic tumor microenvironment. Here we summarize recent progress in our understanding of the role of unfolded protein response activated by endoplasmic reticulum stress in tumorigenesis of pancreatic cancer, and highlight the potential of the cascade of unfolded protein response as therapeutic target for pancreatic cancer.
Keywords
- Endoplasmic reticulum stress
- Unfolded protein response
- Pancreatic cancer
- Apoptosis
Pancreatic cancer is one of the common malignant tumors in the digestive tract. It is extremely difficult to diagnose and treat pancreatic cancer because its clinical symptoms are hidden and atypical, and about 90% of pancreatic cancer are pancreatic ductal adenocarcinoma (PDAC) originating from glandular epithelium [1]. The main reasons for the increase of morbidity and mortality of pancreatic cancer in recent years are the low rate of early diagnosis, high surgical mortality, poor sensitivity to radiotherapy and chemotherapy, and high rate of recurrence and metastasis [2]. Further understanding of the mechanisms that contribute to poor prognosis of pancreatic tumor has great significance for the treatment of pancreatic cancer.
Endoplasmic reticulum stress is a condition in which cells respond to misfolded and unfolded protein aggregation and dysregulation of calcium homeostasis in the endoplasmic reticulum lumen, and activate the unfolded protein response (UPR), endoplasmic reticulum overload response and caspase-12 mediated apoptosis pathway. When the endoplasmic reticulum homeostasis is out of balance, UPR is activated to restore homeostasis by regulating different pathways. UPR has limited regulation ability and adaptability. According to the intensity and duration of the stimulus, UPR can promote cell survival or promote cell death [3]. Recent evidence suggests that the stimulus from tumor microenvironment and toxic chemotherapeutic drugs both cause chronic UPR, which is related to the progression and chemotherapeutic resistance of pancreatic cancer.
Endoplasmic reticulum (ER) is a series of lamellar lumen and tubular lumen composed of membranes in the cytoplasm, which are interconnected to form a pipeline system isolated from the cell matrix. As the largest organelle in the cells, ER can be divided into rough endoplasmic reticulum (RER) and smooth endoplasmic reticulum (SER). ER connects the nucleus, the cytoplasm and cell membrane, and is responsible for the transport of substances from the nucleus to the cytoplasm and cell membrane. ER plays an important role in cell function such as the synthesis of proteins, lipids and sugars. SER also has detoxification functions. For example, SER in hepatocytes contains enzymes that remove fat-soluble wastes and harmful substances produced by metabolism [4].
Recent studies have shown that the complex dynamic structure of ER is largely driven by the process that ER need rearrange its position along the cytoskeleton in order to keep close contact with other organelles, and the force to form and maintain the dynamic structure of ER is regulated by a variety of proteins such as cytoskeleton proteins, endoplasmic reticulum transmembrane proteins [5].
ER is involved in maintaining cell homeostasis and the balance between health and disease [6]. When cells are subjected to certain stimuli such as hypoxia, starvation, infection and environmental changes, unfolded polypeptide chains or misfolded proteins accumulate in ER, and this phenomenon is known as ER stress [7]. In response to ER stress, a series of cascade reactions occur in the cells known as UPR, and UPR determines the fate of cells under ER stress such as autophagy, apoptosis, inflammation, aging, stemness and cell cycle [8].
Abnormal activation of UPR is one important mechanism implicated in many human
diseases. Cell death caused by UPR is mainly related to metabolism,
neurodegenerative diseases and inflammation, while cell survival caused by UPR is
related to malignant tumors [9]. The three UPR sensors activating transcription
factor 6 (ATF6), inositol-requiring enzyme 1
A growing number of studies have shown abnormal long-term UPR activation in pancreatic cancer, which may affect the progression and therapeutic efficacy of pancreatic cancer. Tumor microenvironment and cell signaling cascade play a key role in pancreatic cancer, while UPR is responsible for maintaining cell homeostasis and regulating a variety of signaling pathways that affect cell behavior. Therefore, UPR in pancreatic cancer cells is a potential target to overcome drug resistance of pancreatic cancer [16].
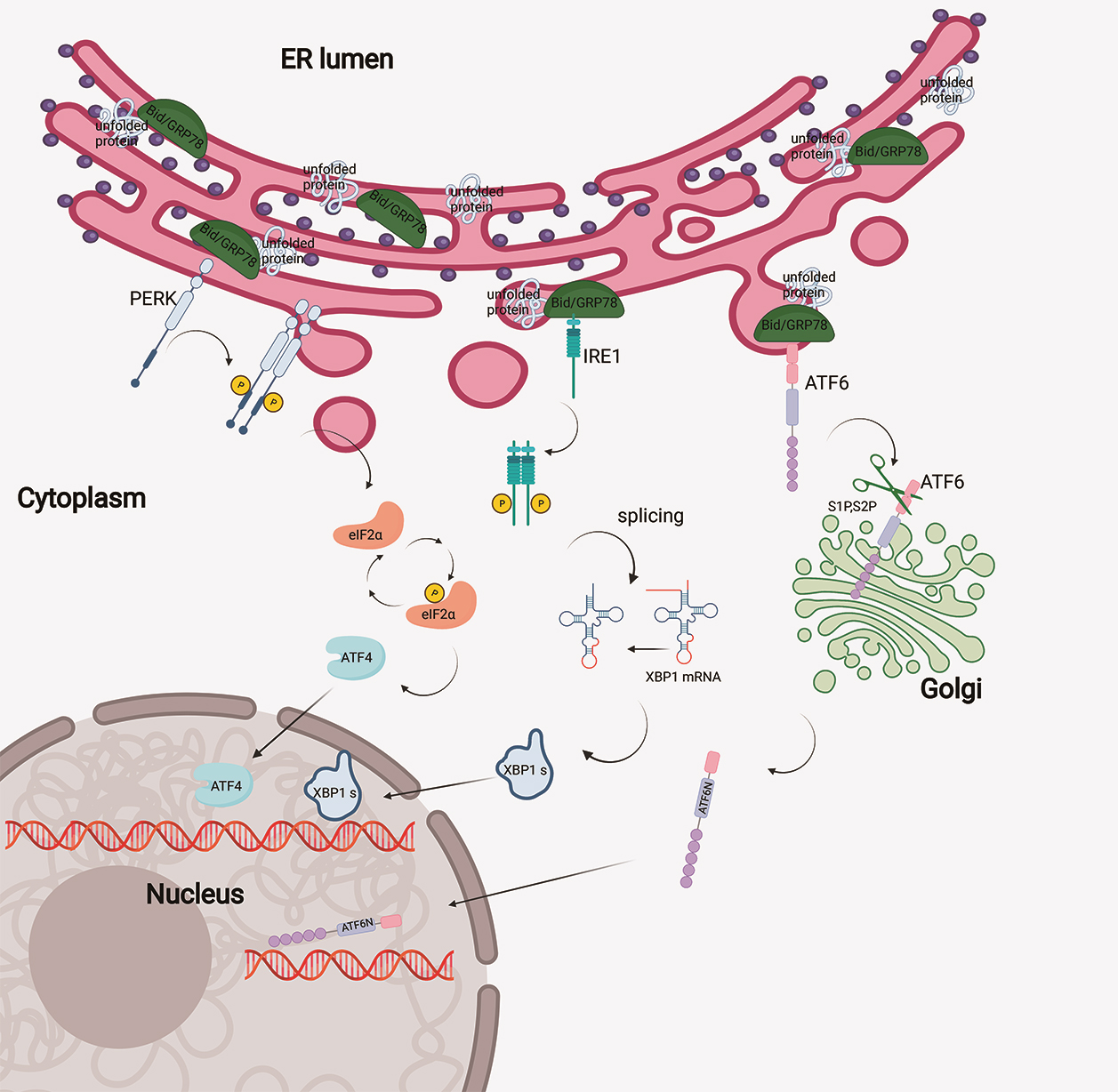
In physiological conditions, the UPR sensors are in an inactive state combined with endoplasmic reticulum resident molecular chaperone protein named glucose regulated protein 78 (GRP78)/binding immunoglobulin protein (BiP). Upon ER stress, GRP78/BiP is activated and the sensors are released [17, 18] (Fig. 1).
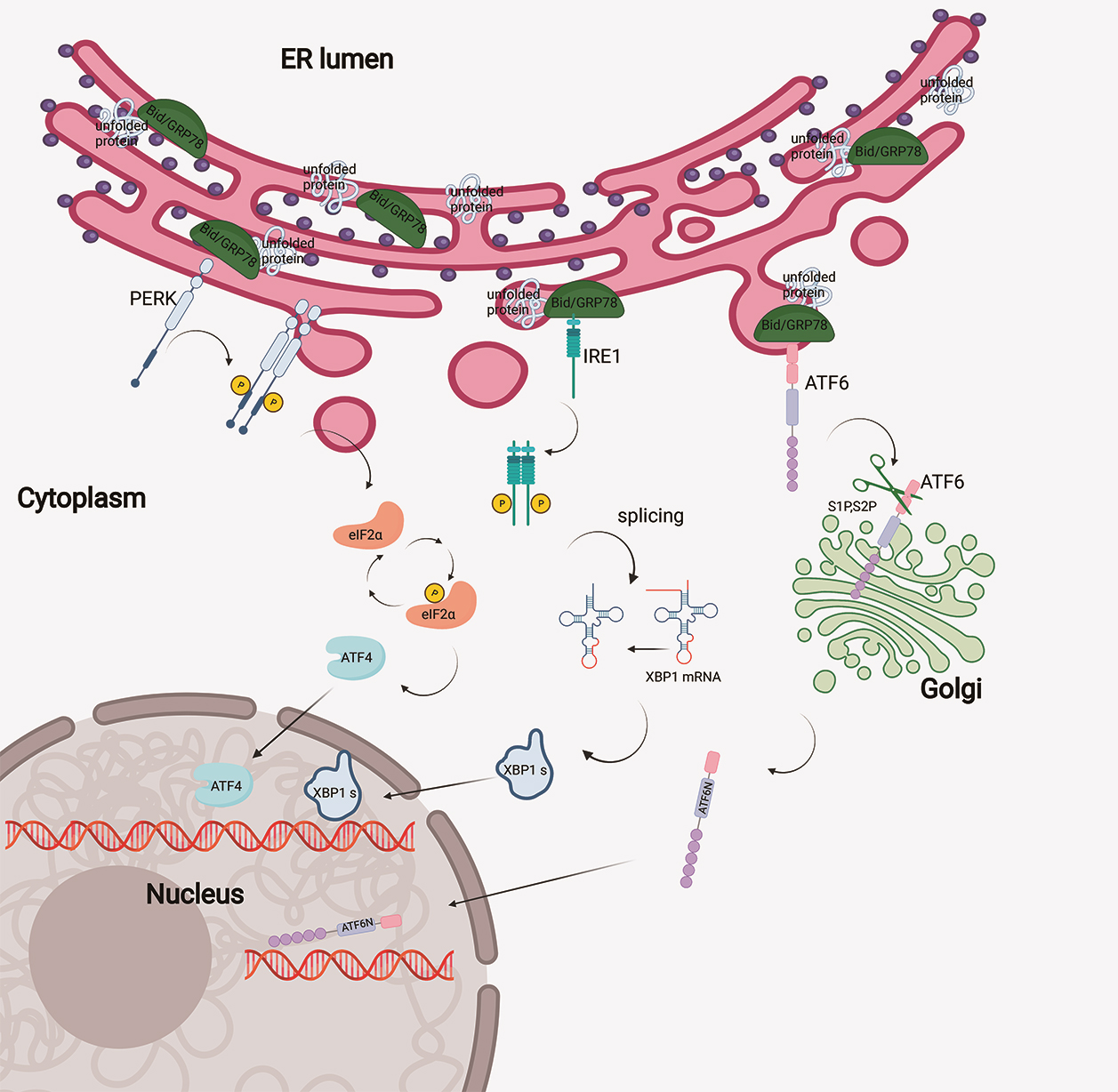 Fig. 1.
Fig. 1.Signaling pathway of the unfolded protein response. Accumulation
of misfolded proteins in ER activates three UPR sensors: PERK, IRE1
In addition, ER stress leads to the release of IRE1 from GRP78, and IRE1 forms a homologous dimer, resulting in self-phosphorylation of Serine 724 and the activation of RNase. The RNase domain of IRE1 has two key functions: processing X Box Binding protein 1 (XBP1) mRNA and regulating IRE1-dependent Decay (RIDD) [19]. IRE1 splices XBP1 mRNA to produce the spliceosome XBP1s. XBP1s is translated as a transcription factor, which can upregulate genes related to ER stress such as ER chaperone [20]. The mechanism by which IRE1 regulates RIDD is not clear, which may be related to the relief of load caused by ER stress by reducing the amount of newly synthesized proteins. In addition to RNase activity, IRE1 domain can stimulate c-Jun N terminal kinase (JNK) signaling pathway by recruiting tumor necrosis factor receptor-associated factor 2 (TRAF2) [21].
PERK is activated in the same way as IRE1 and is self-phosphorylated when
released by GRP78 [22]. Meanwhile, PERK phosphorylates the downstream molecule
eukaryotic translation initiation factor 2A (eIF2
After ATF6 is released from GRP78, two domains of Golgi localization signal are exposed, which are translocated to the Golgi apparatus and cleaved by endopeptidase S1P and endopeptidase S2P to form a fragment ATF6N [27]. ATF6N then enters the nucleus and functions as a transcription factor to induce the expression of XBP1 mRNA and crosstalk with IRE1 pathway [28]. In addition, ATF6N induces the expression of genes involved in ER-associated degradation (ERAD), including ER molecular chaperone such as GRP78, and promotes protein folding [29].
Increasing evidence suggests that the activation of UPR due to hypoxia and nutrient deficiency in PDAC tumor cells may affect the progression and prognosis of PDAC [30]. It is known that complex microenvironment of pancreatic tumor is one of the main reasons for the metastasis of pancreatic cancer and the failure of chemotherapy, radiotherapy and immunotherapy, and microenvironment of pancreatic tumor has attracted more attention. Tumor microenvironment is mainly composed of cancer cells, stromal cells and extracellular matrix components, which affect the signaling cascades responsible for the metabolism and survival of tumor cells [31]. ER plays a key role in protein modification, folding and degradation. However, pancreatic tumor microenvironment disrupts ER homeostasis and affects protein folding, and induces ER stress [32].
The biological characteristics of solid tumors are largely related to the interaction between tumor cells and the microenvironment. Tumor cells are in a state of long-term stress due to high nutrient requirements and insufficient blood supply. Pancreatic tumor cells escape immune surveillance because of unique tumor immunosuppressive microenvironment [33]. Therefore, pancreatic cancer cells can survive under harsh conditions by regulating a series of stress responses [34]. Long-term use of gemcitabine has been shown to lead to extensive reprogramming of the tumor microenvironment, resulting in drug resistance and reduced chemotherapy efficacy in pancreatic cancer. Therefore, understanding the tumor microenvironment and the stress response it induces is important to explore new therapeutic targets [35].
UPR is abnormally activated in pancreatic cancer cells, and determines the survival and death of pancreatic cancer cells [32, 33, 34]. GRP78, as a chaperone protein of ER, regulates a series of signal pathways that promote cell survival and death. Studies have shown that GRP78 is highly expressed in pancreatic cancer cells, and downregulation of GRP78 dramatically affects the metabolism and proliferation of pancreatic cancer cells [36]. In addition, GRP78 is associated with poor prognosis of pancreatic cancer [37]. GRP78 is highly expressed in PDAC cells compared with normal pancreatic duct cells, but how increased expression of GRP78 promotes the progression of PDAC has not been fully explored [38].
On the other hand, PERK activation has been associated with tumorigenesis, and PERK inhibitors have been reported to promote apoptosis in PDAC cells and reduce the size of tumor growth in vivo [39]. An interesting study has shown that the metastasis of pancreatic tumor to the liver, a popular organ for pancreatic cancer metastasis, is associated with selective activation of PERK but not IRE1 [40]. PDAC is characterized by abnormal mucin secretion, which plays an important role in tumor occurrence, metastasis and chemotherapy resistance. GGDPS inhibitor (GGDPSi) could induce the death of PDAC cells by interfering with the transport of mucin, thereby inducing the activation of UPR and apoptosis of PDAC cells [41]. Further studies are needed to reveal the underlying mechanism.
IRE1 and ATF6 activate AGR2 to promote cell proliferation, and they can promote the tumorigenesis of PDAC, while IRE1 inhibitors inhibited PDAC cell proliferation [42]. In addition to ductal cells, PDAC contains acinar cells. Acinar cells have a large demand for protein synthesis, and they are rich in ER and subjected to ER stress and UPR. The loss of XBP1 will lead to the apoptosis of acinar cells [43]. Furthermore, exosomes from pancreatic cancer cells regulated ER stress by activating p38 MAPK pathway to induce T lymphocyte apoptosis and enable pancreatic cancer cells to survive [44].
Taken together, although these studies have demonstrated that major ER stress sensors are activated in pancreatic cancer cells, their activation mechanisms and how they affect pancreatic tumor cell behavior and destiny remain to be investigated.
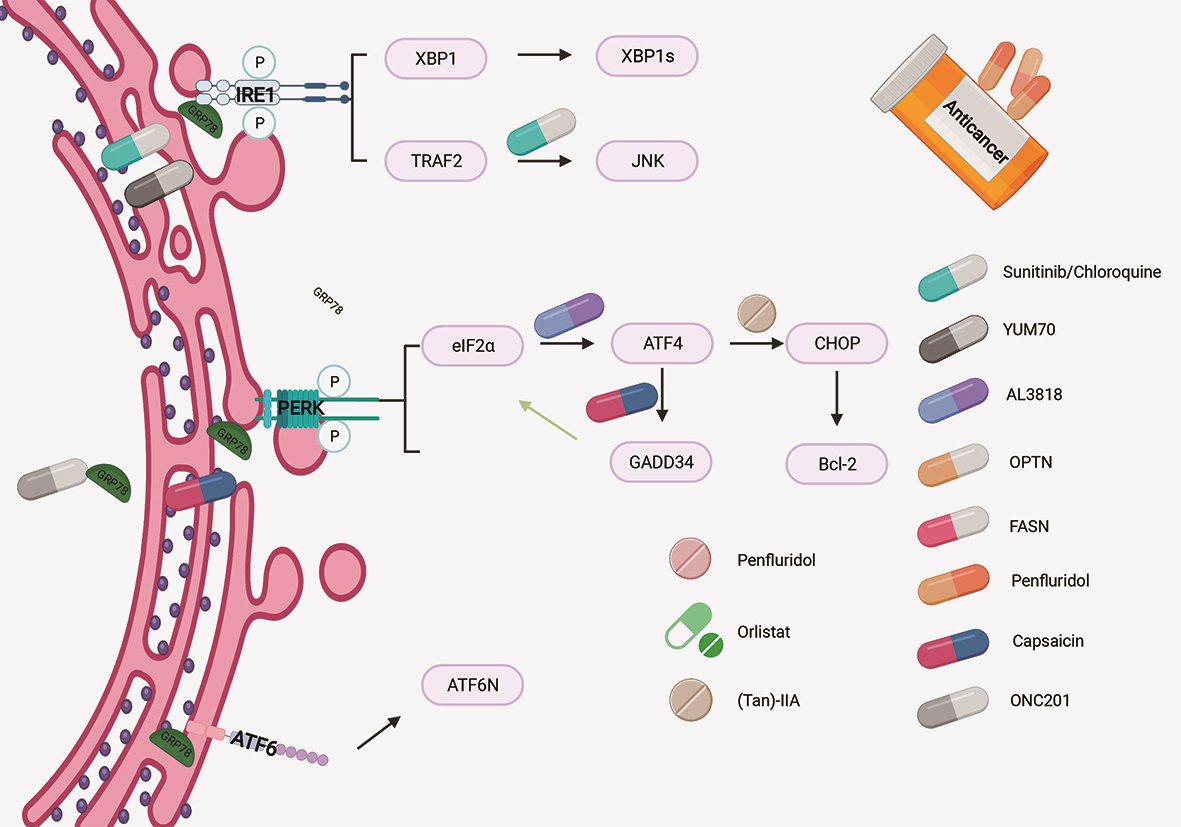
Inhibition of autophagy mediated by UPR can enhance the efficacy of chemotherapy drugs in the treatment of pancreatic cancer [45] (Fig. 2). Thakur et al. [46] showed that in vitro and in vivo PDAC models, the survival rate of cells or animals increased significantly after treatment by the combination of sunitinib and/or chloroquine with gemcitabine, but the toxicity did not increase significantly. Sunitinib combined with chloroquine could inhibit the growth of pancreatic tumor by inhibiting autophagy and increasing apoptosis. These results suggest that stress-UPR-lysosomal pathway can be targeted to improve the treatment of PDAC. Collectively, these results suggest that ER stress mediated autophagy could enhance the efficacy of chemotherapy and could be a novel therapeutic approach for PDAC.
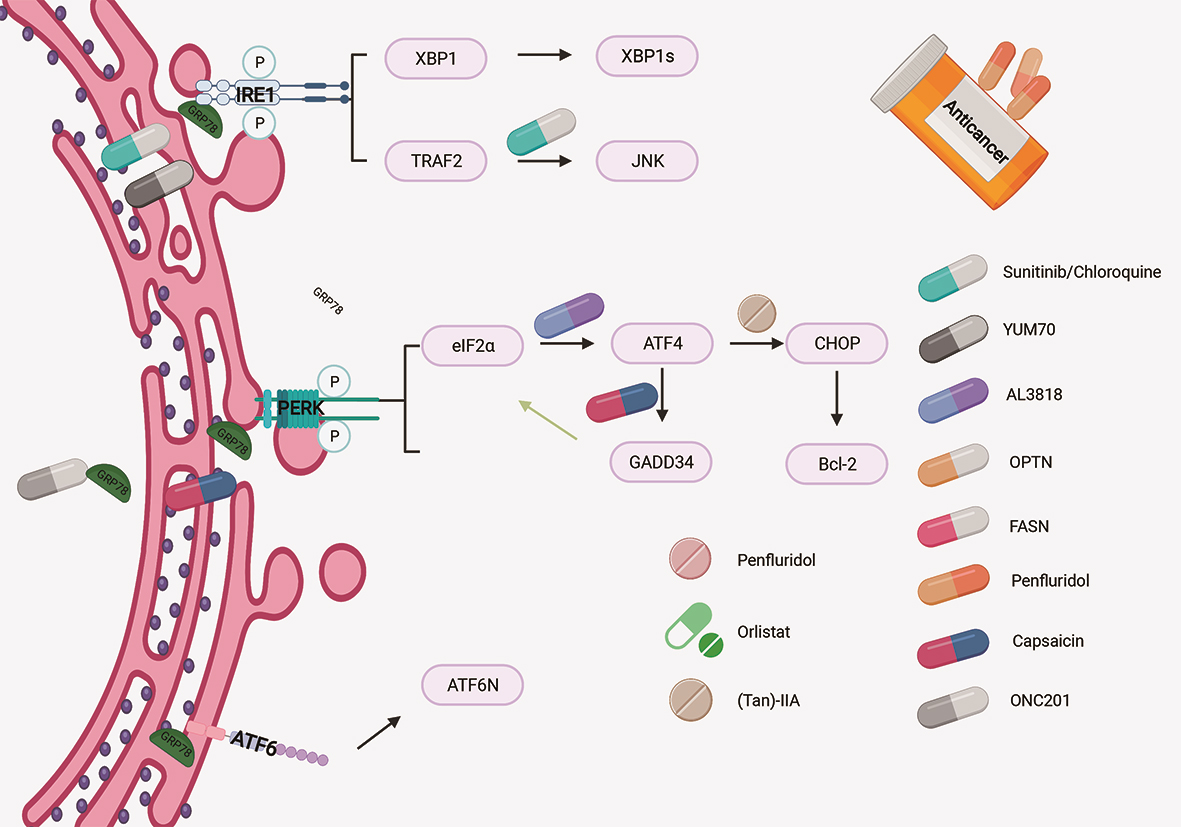 Fig. 2.
Fig. 2.Schematic representation of how the drugs regulate UPR in pancreatic cancer cells.
Hydroxyquinoline analogue (YUM70) directly combined with GRP78 to induce ER stress-mediated apoptosis of pancreatic tumor cells in a pancreatic cancer transplanted tumor model, leading to slow tumor grow and small tumor size, and had no toxic effect on normal tissues [47]. Therefore, YUM70 is a potential drug targeting UPR for pancreatic cancer treatment.
As an anti-tumor drug, Anlotinib (AL3818) inhibits the proliferation of
pancreatic cancer cells, and promotes the apoptosis of tumor cells by
accumulating ROS and causing ERS through PERK/p-eIF2
Optineurin (OPTN) is an important member of the autophagy system and is highly expressed in pancreatic cancer. OPTN knockdown is associated with UPR activation and endoplasmic reticulum chaperon-mediated autophagy. Therefore, OPTN is a potential target for the treatment of PDAC [49].
The expression of fatty acid synthase (FASN) was significantly increased in pancreatic cancer. FASN inhibitor Orlistat had a synergistic effect with gemcitabine in pancreatic cancer cell and orthotopic transplantation model, partly because Orlistat caused decreased cytotoxicity and increased apoptosis related to ER stress. Therefore, regulating fatty acid biosynthesis pathway may overcome gemcitabine resistance in pancreatic cancer by regulating ER stress [50]. Penfluridol induces autophagy of PDAC cells through UPR, which inhibits the growth of pancreatic tumors and provides a new therapeutic target for advanced chemotherapy of pancreatic cancer [51].
Tanshinone (Tan)-IIA, a derivative of Phenan-threnequinone, increased the expression of PERK, ATF6, Caspase-12 and CHOP, resulting in ER stress induced apoptosis of pancreatic cancer cells. In vivo experiments confirmed that (Tan)-IIA inhibited the growth of pancreatic cancer [52].
Capsaicin has shown anti-tumor effect in various cancer cells, effectively
inhibiting the growth and metabolism of pancreatic tumor and prolonging the life
span of mice with pancreatic cancer. In PDAC, Capsaicin promoted the expression
of some key molecules of ER stress pathway, including GRP78, PERK,
phospho-eIF2
ONC201 is a small molecule drug with anticancer activity in clinical trials. ONC212, a fluorinated-ONC201 analogue, has shown anti-pancreatic cancer activity associated with UPR induced by IGF1-R and GRP78/ BIP [54]. Furthermore, RM-581 inhibited pancreatic tumor growth by inducing ER stress mediated apoptosis of pancreatic cancer cells [55].
With deep understanding of the role of ER in physiological and pathological conditions, the restoration of ER homeostasis provides us with a new direction for the development of effective strategies for disease treatment. In response to ER stress, UPR regulates a variety of cellular processes, including the synthesis and degradation of intracellular proteins. Due to the complexity of tumor microenvironment, chronic UPR is activated in tumor cells and affects tumor progression and prognosis. Targeted regulation of ER stress pathway and UPR is expected to be a new and effective method for cancer therapy.
With regard to PDAC, we can induce cancer cell death by inhibiting the activation of pro-survival signaling pathway induced by ER stress. As we all know, although gemcitabine is the main chemotherapy drug of PDAC, the most difficult problem to overcome is drug resistance. Furthermore, some studies have shown that gemcitabine may lead to the recurrence of invasive tumor types [56]. This is one reason for the high recurrence rate and metastasis rate of PDAC. We propose using the inhibitors of UPR pathway in combination with gemcitabine to enhance the chemosensitivity of cancer cells without obvious increase in toxicity, which is a new direction to be explored.
Logue et al. [57] recently reported that chemotherapy drug paclitaxel enhanced the expression of cytokines IL-8 and IL-6 in breast cancer cells in a manner related to IRE1, and this phenomenon could be weakened by IRE1 RNase inhibitors. Indeed, there is crosstalk between UPR and inflammatory signaling pathway in cancer cells so that cytokines are activated through UPR key molecules such as BXP1, GRP78, IRE1 [58].
Last but not least, a recent review summarized the role of noncoding RNAs, in particular miRNAs, in the regulation of ER stress and UPR in cancer cells [59]. It is interesting that noncoding RNAs including miRNAs have been implicated in pancreatic cancer [60, 61]. The identification and characterization of noncoding RNAs that regulate UPR in pancreatic cancer cells will provide another direction for targeting UPR for pancreatic cancer therapy.
DL designed this review and drafted the manuscript. DL and KC searched the related literature and prepared the figures. XT and YY revised the manuscript. All authors contributed to the article and approved the submitted version.
Not applicable.
Not applicable.
This study was supported by Natural Science Foundation of China (No. 81871954, 81672353) and Beijing Municipal Natural Science Foundation (No.7212111).
The authors declare no conflict of interest.