
The epigenetic memory stored in the dynamic modifications, such as base modifications of cytosine (C) in DNA, including methylation/hydroxymethylation/demethylation, causes heritable phenotypes via regulating gene expression without alteration of DNA sequence. The process from cytosine modification to the epigenetic effect is orchestrated by complicated machinery consisting of writers, erasers, readers, and other factors. The two major forms of cytosine modification include methylcytosine (5-mC) and hydroxymethylcytosine (5-hmC). DNA methyltransferases (DNMTs) including DNMT1, DNMT3A, and DNMT3B function as writers for 5-mC. The ten-eleven translocation proteins (TET) including TET1, TET2, and TET3 in the mammalian genome are responsible for hydroxymethylation of 5-mC to generate 5-hmC, 5-formylcytosine (5-fC), and 5-carboxylcytosine (5-caC). The 5-mC and 5-hmC have become the two most extensively investigated epigenetic markers, and the dynamic balance of these two markers shape the landscape of the epigenome, functioning as a platform to regulate gene expression epigenetically. The landscape of the 5-hmC in epigenome is precisely and tightly regulated during the development. Aberrant alterations of the epigenetic regulation may cause severe consequences such as phenotype change as well as initiation of disease. Progressively, significant achievements have been made in characterization of writers, erasers, and readers of 5-mC and 5-hmC, as well as the contribution of aberrant alteration of 5-hmC/5-mC landscape to the pathogenesis of human diseases, such as cancers and neurological disorders. This article will highlight the research advances in the distinct contribution of TET proteins as suppressors or promoters to the pathogenesis of tumorigenesis and progression. Furthermore, this article also discusses the challenges and the directions for research in the future.
A body of evidence has linked aberrant epigenetic regulation of gene expression involved in essential metabolic pathways to distort growth, development, and pathogenesis of diseases. Of the epigenetic modifications identified and characterized so far, base modifications such as methylation, hydroxymethylation, and demethylation of cytosine (C) have been extensively investigated. 5-methylcytosine (5-mC) and its oxidative derivative, hydroxymethylcytosine (5-hmC) in DNA and RNA, have been appreciated as the epigenetic markers crucially involved in the regulation of almost all metabolic pathways for cellular processes [1]. The main components in the machinery driving for methylation and hydroxymethylation of cytosine includes DNMTs as writers, TET family members as erasers, and direct interaction proteins as readers. They have been identified and characterized biochemically, molecularly, genetically and epigenetically. The writers are enzymes responsible for methylation of cytosine, while the three TET family members function as 5-mC erasers for the conversion of 5-mC into 5-hmC and further to 5-formylcytosine (5-fC) and 5-carboxy cytosine (5-caC) (Fig. 1).
 Fig. 1.
Fig. 1.Structure of ten-eleven translocation proteins (TETs) in mammals
and TETs-mediated hydroxymehtylation. (A) TETs, including TET1, TET2, and TET3,
incorporate a catalytic domain (CD) that harbors a double-stranded
5-mC is the most extensively studied epigenetic marker in mammalian genome [2, 3, 4, 5, 6, 7]. Although 5-hmC was discovered decades ago [8, 9, 10, 11, 12, 13, 14], it wasn’t received significant attention until in recent years [13, 15, 16, 17]. 5-hmC serves as an intermediate for demethylation in mammalian brain [13, 15, 16, 17] and ES cells [18, 19, 20]. Furthermore, 5-hmC is a predominant epigenetic marker for shaping epigenome landscape, critically regulating chromatin architecture and subsequent gene expression [21, 22]. For example, 5-hmC loss has become a hall marker for cancer cells [23, 24, 25, 26, 27, 28]. The abundance and landscape of 5-hmC are in a tissue dependent manner [29, 30, 31].
The proteins that specifically recognize the markers 5-mC or 5-hmC have been identified and characterized as the readers. The networks consisting of writers, erasers, and readers cover nearly all the metabolic pathways, regulating all biological processes. The levels and specific distribution of these epigenetic markers in the epigenome and the epitranscriptome are precisely regulated in accordance with the developmental stages such as growth, reproduction, and response to environmental exposure. The regulation of such complicated biological processes is orchestrated during the life span by coordination of the writers, erasers, readers, as well as other factors. Trivial alteration of base modifications may cause devastating damages to metabolic pathway(s), leading to severe consequences such as phenotype changes and initiation of diseases.
There are six members in DNMT family, including DNMT1, DNMT2, DNMT3A, DNMT3B, DNMT3C, and DNMT3L [32]. However, only three members—TET1, TET2, and TET3 in the TET family have been identified and characterized in the mammalian genome [18, 33]. The aberrant methylation and demethylation have been linked to pathogenesis of diseases, such as cancers, neurodegenerative disorders, and others (heart, liver, kidney, lung, and immunological disorders) [34, 35, 36, 37]. Of the main components in the machinery for cytosine modifications, TET family members shape the landscape of the epigenome via regulation and disposition of 5-hmC levels [38, 39] and distinctly function as regulators for brain development as well as suppressors and promoters for tumorigenesis [26, 40, 41, 42, 43, 44, 45, 46] (Tables 1,2,3,4, Ref. [15, 34, 35, 36, 37, 47, 48, 49, 50, 51, 52, 53, 54, 55, 56, 57, 58, 59, 60, 61, 62, 63, 64, 65, 66, 67, 68, 69, 70, 71, 72, 73, 74, 75, 76, 77, 78, 79, 80, 81, 82, 83, 84, 85, 86, 87, 88, 89, 90, 91]).
| Common chemical functions of TET proteins | References |
| Conversion of 5-mC to 5-hmC | [15] |
| Conversion of 5-hmC to 5-fC | [47] |
| Conversion of 5-fC to 5caC | [48] |
| TET | Hydroxymethylation region | References |
| TET1 | Hydroxymethylation of promoter | [49] |
| Transcription start sites | [50] | |
| TET2 | Gene body, Enhancer | [51] |
| TET3 | Hydroxymethylation of gene during embryonic development | [51] |
| Interactions | References |
| TET1 interaction with | |
| GADD45A and NONO | [52, 53, 57] |
| MBD1, MBD3, PRC2 | [54, 55, 56] |
| SIN3A | [57, 58] |
| NANOG | [59] |
| HIF1a, HIF2a | [60, 61] |
| EGR1 | [62] |
| TET2 interaction with PSPC and EP300 | [63, 64] |
| Functional interplay of TET1 and DNMT3A | [65, 66] |
| Interplay between TET2 and DNMTs | [65, 66, 67] |
| Interaction with regulatory elements | [34, 54, 57] |
| TET3 interaction with thyroid hormone nuclear receptors (stabilizes their association to chromatin) | [68] |
| Interaction with O-GlcNAc transferase (OGT) | [69] |
| Interaction with NSD3 and REST | [70] |
| TET | Physiological functions | References |
| TET1 | Maintenance of pluripotency of stem cells | [71, 72] |
| Differentiation | [73, 74] | |
| TET2 | Reprogramming | [75, 76, 77, 78, 79] |
| Diabetic skin diseases | [80] | |
| Coronary heart disease | [81] | |
| NK cells maturation | [34] | |
| Allergic rhinitis | [35] | |
| TET3 | Demethylate the genome of the fertilized zygote to allow it to grow into a fully developed organism | [82] |
| Neuron development and maturation | [83] | |
| Neuron cells repair and recovery | [84] | |
| Neural progenitor cells specification/cellular identity/Proliferation/Differentiation | [36, 85, 86] | |
| TET3-deficiency syndrome and Mendelian inheritance patterns | [86] | |
| Bone marrow hypoxia and development of Xenopus tadpole brain | [87, 88] | |
| TET1/TET2/TET3 | Critical for brain functions | [85, 86, 89, 90, 91] |
| Mutations or changes are associated with/the cause of cognitive deficits in human | [36] |
This review article briefly explores the expression patterns and physiological functions of TETs, and mainly focuses on the recent advances in the distinct functions of TET proteins in the regulation of tumorigenesis. The challenge and direction for research in the future are discussed.
Demethylation of the constitutionally methylated promoters of some oncogenes,
such as protooncogene
Initially, it was accepted that TET1 was a direct or indirect tumor repressor [92]. Indeed, repression of TET1 expression or aberrant subcellular localization of TET1 in gastric cancer (GC) tissues led to carcinogenesis associated with significantly lower 5-hmC, suggesting the crucial role of TET1 as a cancer repressor [93]. However, this conclusion was challenged further by the fact that the upregulation of TET1 expression was also detected in some cancers. However, more emerging evidence has disputed gradually the original controversy regarding TET’s role in tumorigenesis and progression. Based on current knowledge, any of the three TET members could repress and promote tumorigenesis in a cancer type-dependent manner. However, TETs’ function on cancer suppression or promotion relies on activation or inactivation of crucial genes involved in signaling pathways for oncogenesis or anti-tumorigenesis via direct hydroxymethylation (not demethylation alone) and/or interaction with other factors [94]. In the case of TET1 as a tumor suppressor, TET1 expression was downregulated and completely repressed in human cancer samples or cell lines (Fig. 2, Ref. [95]).
 Fig. 2.
Fig. 2.TET1 functions as a tumor suppressor. (A) In normal cells, TET1 expression is maintained at a regular level. TET1 is recruited to the methylated promoter area of the tumor suppressor genes for demethylation so that the tumor suppressor could be expressed at normal levels. (B) Due to the mutation of Tet1 genes and other factors, Tet1 expression was silenced, leading to the silencing of the suppressor genes and activation of the oncogenes, such as Ezh2. Expression of the oncoproteins initiates tumorigenesis or enhances cancer cell growth and metastasis (Cited and modified from Ma et al., Cytosine Modifications and Distinct Functions of TET1 on Tumorigenesis. Chromatin and Epigenetics Eds. By Dr. Colin Logie and Dr. Tobias Knoch, The Netherland 2019. Intechopen.83709 http://dx.doi.org/10.5772/intechopen.83709) [95].
It has been well-known that miRNAs are involved in tumorigenesis or anti-tumorigenesis, functioning as oncogenic miRNAs or anti-tumor miRNAs. This mechanistically refers to the oncogenic miRNA-mediated repression of TET or other tumor suppressors. Recently, it was demonstrated that lnc-RNAs and circular RNAs (circRNAs) were also involved in tumorigenesis and progression via silencing of TET1 expression.
TET1 has been implicated for interplay with some anti-tumor miRNAs. For example, TET1 silencing led to significant down-regulation of miR-7 and miR-15/16 targeting to proto-oncogenes CdkK6, Mcf2L, and Edn1 and Wnt4 as well as Fzd4 components in the Wnt signaling pathway in papillary thyroid carcinoma (PTC). Consequently, the expression of these proto-oncogenes and the Wnt pathway was dramatically upregulated [96], and accordingly the cell proliferation and invasion were significantly enhanced. This suggests TET1 as a tumor suppressor for PTC and as a potential target for therapy of PTC patients [96].
TET1 has been characterized to interplay with anti-tumor lnc-RNA as well. As an example, the downregulation of lnc-01089, a lnc-RNA in GC tissues and cell lines caused an upregulation of miR-27a-3p and the subsequent repression of TET1. These molecular events led to the enhanced proliferation, migration, and invasion of GC cells. Associated phenotypes include larger tumor size, higher T-stage, and lymphatic metastasis in GC patients. Consistently, the lnc-01089 overexpression reversed all the phenotypes conferred by the downregulation of this lnc-RNA. Thus, the lnc-01089-miR-27a-3p-TET1 could potentially become therapeutic targets for GC [97].
TET1 functions as an indirect tumor suppressor and promoter in different cancers via interplay with some miRNAs that bear dual functions on promotion or repression of some cancers. For example, miR-34a could function as tumor suppression for lung tumorigenesis but as tumor promotion for breast cancer. It has been shown that 3,6-Dihydroxyflavone (DHF) activates miR-34a expression via elevation of TET1, which directly binds to its own promoter region [56] to regulate 5-hmC landscape. While DHF does not alter the binding of TET1 to its own promoter region, it could inhibit DNMT1 activity and hypermethylation, leading to enhanced demethylation of the TET1 promoter and activation of TET1 expression. Consequently, the elevated expression of TET1 enhances demethylation of the miR-34a promoter, leading to its activation and subsequent breast carcinogenesis [98]. However, miR-34a is crucially involved in repressing tumor progression through regulating epithelial-mesenchymal transition (EMT) via EMT-transcription factors, p53, and other essential signaling pathways [99].
TET1 is also involved in the circRNA-mediated epigenetic regulation of human retinoblastoma (HRB). The dynamic alteration of circRNAs was detected in HRB in in ways that circRNAs with increased expression, decreased expression, and unchanged expression relative to these in the adjacent tissue. Of the down-regulated circRNAs, some of their host genes were involved in chromatin modifications, such as TET1-has_circ_0093996 that takes TET1 as its host gene in HRB. The downregulation of the TET1-has_circ_0093996 was associated with the decrease of programmed cell death 4 (PDCD4) in the primary and recurrent HRB samples. Further bioinformatic study has established the complete regulatory axis TET1-has_circ_0093996-miR-183-PDCD4, suggesting the circRNA-based regulation on RB pathogenesis.
In addition to interaction with anti-tumor miRNAs, TET1 can also interplay with some miRNAs that could enhance tumorigenesis. For example, upregulation of miR-29a-5p accompanied by activation of the IL6-STAT3 pathway downregulated the expression of TET1 and TET3 consistently with lower levels of 5-hmC in colitis-associated colorectal cancer (CAC). In vitro knockdown of miR-29a-5p in cell lines could reverse the molecular events in TET and the IL6-STAT3 pathway. Thus, it could be inferred that the miR-29a-5p/STAT3/TET signaling axis may contribute to initiation of CAC and potentially serve as targets for therapy or prevention of CAC [100].
TET1 upregulation leads to hypomethylation of the BRCA1 promoter and upregulation of the DNMT3 confers hypermethylation of Synuclein Gamma gene (SNCG). Enhanced expression of TET1 is associated with down-regulation of miR-29b, suggesting Tet1 as a target of miR-29b. Therefore, the potential of the drug that bears the dual function on the regulation of promoter methylation/demethylation, may potentially become a promising therapeutic option for some cancers [101]. Similarly, overexpression of miR-29b associated with the downregulation of TET expression promotes cell proliferation, colony formation, migration, and EMT, suggesting miR-29b as a BC/GC promoter [102, 103]. Likewise, miR-21-5p and miR-4284 target and silence Tet1 expression in colorectal cancer (CRC) and GC SGC-7901 cells, respectively. Thus, both miR-21-5p and miR-4284 may serve as oncogenic biomarkers for diagnostics and prognostics in CRC and GC [104, 105].
Chronic inflammation leads to epigenetic alteration, in particular the
methylation/demethylation mediated by DNMTs and TETs. Human gingival fibroblast
exposure to IL-1
The
Another example is the silencing of TET1 expression mediated by the
Wnt/
The epithelial-mesenchymal transition (EMT)-related oncogene pathway is silenced by TET1. TET1 promotes stemness properties in cervical cancer and inhibits EMT through 5hmC-dependent and -independent mechanisms. TET1-dependent elevation of 5hmC levels in the promoter regions of the ZEB1 and VIM genes doesn’t activate the expression of both genes. Instead, both genes were silenced. The silencing of the Zeb1 and Vimgenes attributes to loss of histone H3K4 and gain of histone H3K27 via interaction of TET1 with LSD1 and EZH2, the two histone modifiers. LSD1 and EZH2 are recruited by TET1 to the Zeb1 promoter, subsequently silencing Zeb1, an ETM-related oncogene [110, 111].
TET1 is silenced by EZH2 and H3K27me3 in triple-negative breast cancer (TNBC) cells, which lack the expression of ER, PR and HER2. High levels of EZH2 and low levels of TET1 in TNBC patients are associated with low survival rate. EZH2 downregulates TET1 expression via epigenetic regulation of H3K27me3, thereby blocking the p53 signaling pathway and leading to tumor progression. Consistently, knockdown of EZH2 with specific inhibitors GSK343 or shRNA, could upregulate TET1 and subsequently activate p53 pathway. It could also stimulate cell cycle arrest and senescence in TNBC cells. Thus, the EZH2-H3K27me3-TET1 axis could become a potentially promising therapeutic target via modulation of the epigenetic landscape [112].
Epidermal growth factor receptor (EGFR)-mediated TET1 repression promotes growth of some cancer cells, such as lung adenocarcinomas and glioblastomas HCC827/Del and HCC827/Del-T790M cell line. This suggests that TET1 may serve as a therapeutic target for lung cancers and glioblastomas both induced by oncogenic EGFR [113]. However, in human non-small cell lung cancer (NSCLC) patient samples, there was no EGFR-mediated TET1 silencing. Instead, a significant elevation of the TET1 expression level was detected in the patient tumor tissues with EGFR mutations. Thus, further studies are indispensable to explain the discrepancy between cancer cell lines and human cancer tissues [114].
A recent study discovered that oxidative stress leads to TET1 suppression and tumorigenesis. Treatment of mouse liver cells with cadmium (Cd) stimulated cell invasion. Molecularly, Cd treatment-induced oxidative stress, associated with overproduction of oxidative stress-sensitive metallothionein 2A (MT2A), led to suppression of TET1 and ApoE, but no effects were seen on DNMTs. Additionally, Cd could downregulate the expression of tissue inhibitors of metalloproteinase 2 and 3 (TIMP2 and TIMP3). The expression of ApoE, TET1, TIMP2, and TIMP3 was further suppressed by hydroquinone, a ROS species. However, the suppression was reversed by N-acetyl-l-cysteine (NAC), a ROS scavenger. Excitingly, the cell invasion induced by Cd was abolished by NAC. Therefore, the suppressed expression of TET1, ApoE, TIMP2, and TIMP3 by Cd and the induced ROS was likely involved in the enhanced invasiveness in response to Cd exposure. Anti-ROS could reverse the toxicity in terms of the Cd-induced malignancy [115].
TET1 is also silenced by the deficiency of key genes involved in base excision repair (BER). 5-mC/5-hmC levels were investigated in the 3-methylcholanthrene (3-MCA) induced cell malignant model, rat chemical carcinogenesis model, and human lung cancer tissues, respectively. The expression of XRCC1, OGG1, and APEX1 genes were crucial for the BER pathway and was significantly downregulated in response to 3-MCA treatment. Meanwhile, the expression of TET1 itself was suppressed by DNA methylation. Restoration of TET1 expression reversed the proliferation, migration, and invasion in the 3-MCA-induced malignant cells. Thus, TET1 could serve as a potential biomarker and therapeutic target for lung cancer [116].
Several natural or synthetic small molecules, such as eicosapentanoic acid
(EPA), ML309, vitamin C (VC), chrysin, luteolin, and resveratrol (Res), have been
identified to upregulate the expression of TET1 in cancer cells. In
hepatocarcinoma cells, one of the major polyunsaturated fatty acids, could
enhance rapid demethylation via the formation of
PPAR
Downregulation of TET1 inhibits AJAP1 expression via the reduction of 5-hmC
levels at the promoter of adherens junctions associated protein 1 (AJAP1),
subsequently activating
IDH1 gene mutation leads to the generation of 2-hydroxyglutarate
(2-HG), an oncometabolite that inhibits the activity of
Treatment of GC with chrysin, a natural compound frequently present in honey, could significantly upregulate the expression of TET1 and the subsequent elevation of 5-hmC in GC MKN45 cells, thereby inducing cell apoptosis and blocking cell migration/invasion. Thus, the anti-tumor effects of chrysin was mediated through upregulation of TET1 expression in GC, suggesting TET1 as a potentially novel target for GC therapy [119].
Luteolin, a dietary flavone, modulates numerous signaling pathways involved in carcinogenesis such as colon cancer via the downregulation of DNMTs, upregulating TET1 expression and enhancing recruitment of TET1 to the Nrf2 promoter region for hydroxymethylation to promote Nrf2 expression. In addition, the interaction of TP53 and Nrf2 enhances apoptosis. Therefore, this discovery not only partially elucidates the anticancer mechanism of luteolin but also suggests Nrf2 and Tet1 as the potential therapeutic targets for colon cancer and possibly other cancers [120].
Gastric adenocarcinoma (GA) with enteroblastic differentiation (GAED), a rare variant of aggressive adenocarcinoma, is frequently associated with mutation-caused TP53 inactivation. Of the characterized GAED cases, nearly 60% performed TET downregulation accompanied with the declination of 5-hmC levels, which is inversely associated with advanced stage, tumor size, and lymph metastasis. Approximately 70% of the cases with hypermethylation of the TP53 promoter is associated with the downregulation of TET1 and the reduced 5-hmC levels. Thus, it suggests that the promoter hypermethylation of TP53 caused by decreased levels of TET1 and 5-hmC partially contributes to loss of TP53 expression, further suggesting TET1 and TP53 as tumor suppressors and potential therapeutic targets for GA [121].
Resveratrol (Res), a natural compound, has been detected to suppress the migration and invasion of prostate cancer cells (PCCs) via the upregulation of TET1 expression. This enhanced the hydroxymethylation and expression of TIMP2 and TIMP3 but suppressed the expression of MMP2 and MMP9. Thus, the Res-mediated inhibition effect on the migration and invasion of PCCs was thought to be via the TET1/TIMP2/TIMP3 pathway, suggesting that this pathway may potentially be employed as a target for therapy [122].
Tet1 has been acknowledged as a tumor suppressor or oncogene depending on the cancer types. Osteosarcoma cells (U2OS) treated with hydroxyurea, an anti-cancer agent, led to TET1 upregulation associated with elevated levels of 5-hmC, enhanced apoptosis, inhibition of cell proliferation, and alteration of cell cycle, suggesting that TET1 could function as suppressor for osteosarcoma [123].
To identify new potential prognostic markers for head and neck squamous cell carcinoma (HNSCC), the promoter methylation status of five neuropeptide receptor genes was examined, including NTSR1, NTSR2, GHSR, MLNR, and NMUR1. Of the detected methylation status in the promoters of five genes, methylation status of GHSR and NMUR1 promoters could be utilized to predict recurrence in HNSCC independently. Combined data analysis of the methylation status of TET family genes indicated a trend toward greater methylation indices as the number of TET methylation events increased [124].
Chemoresistance is defined as the resistance of cancer stem cells (CSCs) or tumor initiating cells to a specific therapeutic agent. It refers to the resistance of a particular tumor to chemotherapy caused by DNA repairability, slower cell cycles, and multidrug resistance genes (MDR) such as P-glycoprotein (P-gp). The chemoresistance contributes to the setup of obstacles against chemotherapy. Of the resistance mechanisms characterized so far, TET-mediated epigenetic regulation plays a crucial role in the development of cancer progenitor cells invulnerable in exposure to conventional cancer therapies.
TET1 deficiency significantly contributes to gemcitabine resistance in cholangiocarcinoma (CCA) cell lines. Relative to normal CCA cells, TET1 is almost silenced, while expression of the P-gp encoded by one of the MDR genes is significantly upregulated. Consistently, TET1 overexpression significantly enhances the sensitivity of CCA cells to gemcitabine, in contrast to the dramatic downregulation of the P-gp expression in gemcitabine-resistant CCA cells. Altogether, it suggests TET1 as a potentially promising target to overcome the chemoresistance in CCA [125].
Unlike in CCA cell lines, TET1 enhances the progestin-based chemoresistance via the upregulation of expression of the glyoxalase I gene (GLOI) mediated by its promoter hydroxymethylation in type I endometrial cancer. The TET1-mediated progestin resistance was reversed by metformin treatment through the downregulation of the GLOI expression, further suggesting the contribution of the TET1-5hmC-GLOI signaling pathway to progestin resistance in endometrial cancer [126].
Whenever TET1 is crucially involved in activation of oncogene(s), oncogene pathways, or maintenance of cancer stem cells, it will become the compliance of oncogenesis and progression. For example, under normal conditions, oncogene mRNAs are sequestrated by certain anti-tumor microRNAs such as miR-26 by complementing 3’UTRs of its target genes. Unfortunately, miR-26a also targets Tet1 mRNAs. In the case of Tet1 overexpression, miR-26a will be sequestrated by Tet1 mRNA, leading to miR-26 deficiency for silencing its target Ezh2, an oncogene, and thereby activating the oncogene(s) and triggering tumorigenesis (Fig. 3). In addition to miRNAs, other factors have been characterized to contribute to TET1-mediated oncogenesis and progression, such as oxidant stress, the toxicant-dependent pathway, hydroxylation-based activation of multiple oncogenic pathways, p53 loss of function, etc.
 Fig. 3.
Fig. 3.Anti-tumor microRNAs, such as miR-26, are sequestrated by their target mRNAs such as Tet1 mRNAs due to over-transcription of Tet1 gene in tumor cells, leading to miR-26 deficiency for silencing of its oncogene target Ezh2, and thereby activating the oncogene(s) to trigger tumorigenesis. (A) Under normal conditions, Tet1 expression is maintained at very low levels to maintain the anti-tumor microRNAs such as miR-26 at sufficient levels for silencing of the oncogene(s), such as Ezh2. (B) In the case of dramatically enhanced transcription of Tet, miR-26 will be sequestrated by targeting the 3’UTR Tet1 mRNAs, its target gene(s), leading to miR-26 deficiency for silencing Ezh2, its target oncogene. Consequently, the miRNA-mediated expression repression of the oncogenes is released, caused by miR-26 deficiency facilitating the initiation of tumorigenesis. (Cited and modified from Ma et al., Cytosine Modifications and Distinct Functions of TET1 on Tumorigenesis. Chromatin and Epigenetics Eds. By Dr. Colin Logie and Dr. Tobias Knoch, The Netherland 2019. Intechopen.83709 http://dx.doi.org/10.5772/intechopen.83709) [95].
It has been demonstrated that TET1 is a tumor promoter via oxidant stress. Triple-negative breast cancer (TNBC), unlike the regular ER-based BC, is a subtype of BC caused by cancer stem cell-like cells (CSC), irrelevant to estrogen receptors, progesterone receptors, and HER2. TET1 and 5hmC are required to maintain the CSCs in TNBC via hydrogen peroxide-dependent activation of a novel gene cascade involved in self-renewal and propagation of CSCs. Intrinsically, the accumulation of hydrogen peroxide stimulated the downregulation of antioxidant enzyme expression in TNBC cells. Exogenously, systemic inflammation and oxidative stress associated with obesity could act similarly as hydrogen peroxide. Downregulation of antioxidant enzyme expression both endogenously and exogenously contributes to the initiation of CSCs, pathogenesis of TNBC and its recurrence. Given the promotion of TNBC mediated by TET1 involved in the oxidant stress-dependent pathway is associated with obesity, TET1 and its components crucial for the oxidant stress pathway could potentially serve as therapeutic targets for TNBC prevention and therapy [127].
TET1 serves as a tumor promoter via the toxicant-dependent pathway. As a persistent organic pollutant in the environment, perfluorooctanoic acid (PFOA) has been implicated in hepatocellular steatosis via epigenetic alterations and abnormal mRNA splicing. High dose of PFOA downregulated global DNA methylation via the reduced expression of DNMT3A as well as the significantly enhanced expression of TET1, while no substantial change was observed for other members of DNMTs and TETs families. Expression alteration of these epigenetic factors significantly elevated the expression of a subset of genes crucially involved in cell cycle regulation, anti-apoptosis, the mTOR signaling pathway, and certain splicing factors. The protein instead of mRNA levels of multiple splicing factors were significantly declined in response to PFOA exposure, leading to the alteration of the splicing patterns of their target genes. This discovery helped gaining insights into the epigenetic mechanisms of hepatotoxicity caused by PFOA, contributing to the elucidation of pathogenesis and to the development of the potential therapeutic strategy [128].
The interaction of TET1 with HIF1a renders TET1 as a promoter for cancer cell
invasion. Indeed, generation and accumulation of the dysfunctional adipose
derived from human obesity could induce hypoxia, leading to upregulation and
stabilization of HIF1
Under hypoxia conditions, the expression of TET1 was significantly upregulated. TET1-ChIP-seq identified its binding loci in the genome of colon cancer cells. TET1-E2082K, one of the two typical mutations, was identified in the Tet1 gene from more than 100 tumors and adjacent tissues, blocking cell migration enhanced by wild-type TET1. Consistently, downregulation of TET1 repressed the aberrant upregulation of gene expression under the hypoxia microenvironment in tumors, suppressing the migration activity of tumor cells. This discovery is potentially promising for TET1 as a therapeutic target for colon cancer [130].
The hydroxymethylation-based activation of multiple oncogenic pathways by TET1
promotes tumorigenesis. TET1 expression levels were inversely correlated with
survival in advanced-stage epithelial ovarian carcinoma (EOC) but positively
correlated with cell migration, anchorage-independent growth, cancer stemness,
and tumorigenicity. Upregulation of TET1 expression enhanced hydroxymethylation,
leading to the activation of multiple oncogenic pathways (Fig. 4), such as the
Casein Kinase II subunit alpha (CK2
 Fig. 4.
Fig. 4.Activation of the oncogenes via TET protein mediated hydroxymethylation of the methylated promoter regions in the oncogenes, thereby TET functioning as tumor promoters. (A) Under normal conditions, hypermethylation occurs at the promoter regions of the oncogenes, leading to their silencing. (B) In case TET proteins are recruited by their interaction partners to the hypermethylated promoter regions of the oncogenes, hydroxymethylation will be conducted by TET proteins, thereby activating the oncogenes and consequently leading to tumorigenesis. (Cited and modified from Ma et al., Cytosine Modifications and Distinct Functions of TET1 on Tumorigenesis. Chromatin and Epigenetics Eds. By Dr. Colin Logie and Dr. Tobias Knoch, The Netherland 2019. Intechopen.83709 http://dx.doi.org/10.5772/intechopen.83709) [95].
TET1 as a lung cancer promoter is independent of its demethylation activity as well but dependent on p53 loss of function. TET1 was identified and characterized as an oncogene in lung cancer evidenced by the fact that Tet1 KD inhibited lung cancer cell growth and led to the reprogrammed transcriptome independent of its demethylation activity. Expression of oncogene Tet1 was repressed by wild-type p53 via the binding to the Tet1 promoter in non-lung cancer cells. Accordingly, Tet1 gain of function following mutation-based loss of p53 suggests TET1 could become a therapeutic target for lung cancers [132] (Fig. 5).
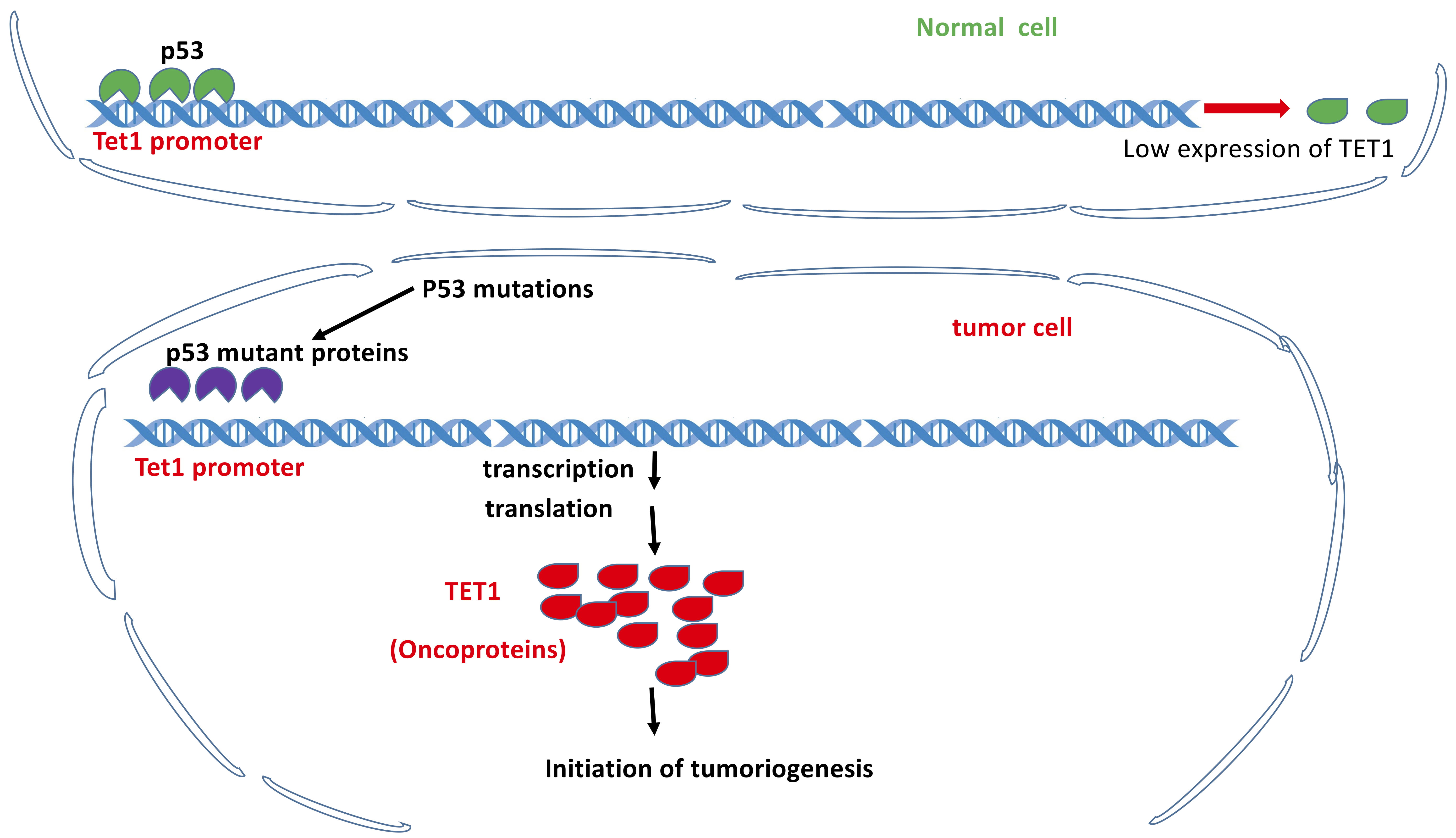 Fig. 5.
Fig. 5.TET1 as a tumor promoter independent of its demethylation activity but dependent on p53 loss of function. Under normal conditions, p53 binds to the promoter of Tet1 gene to maintain the low expression level of TET1 protein. Under the conditions of p53 loss of functions due to mutations, TET1 is overexpressed and functions as tumor promoter.
Several factors contribute to functional TET2 as a direct or indirect repressor, such as hydroxymethylation-independent tumor suppression, inflammatory stress, the interplay between TET2 and some miRNAs, the interplay between TET2 and DNMTs, etc. Downregulation of TET2 expression was detected in nasopharyngeal carcinoma (NPC) cells, while TET2 overexpression suppressed proliferation and invasion of the NPC cells. Interestingly, the effect of TET2 on the NPC phenotype was independent of TET2’s catalytic activity, suggesting the non-catalytic domain function of TET2 in tumor suppression. Molecularly, TET2 repressed proliferation and invasion via the suppression of glycolysis in NPC cells through the interaction of the TET2 N-terminal domain with PKM in the cytoplasm as well as the subsequential prevention of PKM dimers from translocating into the nucleus. Furthermore, TET2 KO performed opposite to TET2 overexpression in terms of NPC progression and survival rate in the xenograft tumor model. Therefore, both TET2 and PKM could potentially serve as targets for NPC therapy [133].
Inflammatory stress significantly contributed to the promotion of the initiation
of pre-leukemic conditions and the aberrant expansion of myeloid cells associated
with Tet2 deficiency. Importantly, antibiotic treatment could reverse the
expansion of Tet2-deficient myeloid and lymphoid tumor cells in
vivo promoted by inflammatory stress. Significant alteration in the expression
of genes involved in the TNF-
Analogous to TET1, TET2 functions as a tumor suppressor via the interplay with some miRNAs. Overexpression of miR-210 in the lung cancer (LC)-derived exosomes was believed to enhance the angiogenesis levels in cancer-associated fibroblasts (CAFs), thereby initiating the CAF proangiogenic switch. MiR-210 contains numerous targets including Tet2 and some repressor genes for the JAK2/STAT3 signaling pathway. Therefore, its overexpression could downregulate TET2 expression and activate the JAK2/STAT3 signaling pathway, thereby facilitating the expression of some proangiogenic factors such as MMP9, FGF2, and VEGF. Consequently, it led to the proangiogenic switch of CAFs [135].
The miR-660-5p was reported to contribute to the pathogenesis of breast cancer (BC), lymph node metastasis, advanced TNM stage, and vascular invasion via the targeted downregulation of TET2 expression. The significant upregulation of miR-660-5p in BC was associated with the downregulation of TET2 and activation of the PI3K/AKT/mTOR signaling pathway, promoting BC cell proliferation and metastasis. In contrast, silencing of miR-660-5p reversed the promotion of BC. Therefore, miR-660-5p mediated promotion of BC progression, partially attributing to modulation of TET2 and PI3K/AKT/mTOR signaling, indicating miR-660-5p/TET2 as a promising therapeutic target for BC [136].
Some environmental pollutants may enhance the interplay of TET2 and DNMTs,
resulting the downregulation of TET2 expression. Bisphenol A (BPA) and its
homolog BSP, the environmental contaminants targeting disruption of estrogen
receptors (ERs), have been implicated in the promotion of breast cancer via the
ER
5-Aza, a DNA methyltransferase inhibitor (DNMTI), has been employed for clinical therapy for the higher-risk myelodysplastic syndromes (MDS). TET2 loss-of-function mutations lead to hypermethylation and impairs the expression of a subset of genes involved in erythroid differentiation in erythroleukemia cells. As one of the mechanisms, 5-Aza could reverse the molecular events induced by the TET loss of function, potentially suggesting the increased efficacy of 5-Aza in MDS patients harboring TET2 loss-of-function mutations [138].
Physical interaction of TET2 with the hydroxymethylated promoters of the signal
transducer and activator of transcription factors (STATs) family members
renders TET2 as a tumor suppressor. In the IFN-
Down-regulation of the ZEB1-mediated TET2 expression could contribute to the pathogenesis of glioma. The expression levels of ZEB1 were significantly increased in the 42 of primary glioma tissues relative to the normal adjacent tissue samples. ZEB1 bound to the Tet2 promoter region led to repression of TET2 expression. Although the detailed mechanisms of how ZEB1 bound to the Tet2 promoter in glioma remains to be elusive, the current study suggests the downregulation of TET2 expression mediated by ZEB1 could contribute to the pathogenesis of glioma. This could shed a light on translation to clinical therapeutic strategies for glioma [140].
TET2 also functions as a suppressor for acute lymphoblastic leukemia (ALL) in children.
Low expression of TET2 in children with ALL is usually associated with poor prognosis. Thus, it was proposed that the TET2 expression levels could serve as a marker for risk assessment of 5-year overall survival (OS) and event-free survival (EFS) [141].
Natural small molecules functional as activators for TET2 activity could have anti-tumor functions, such as ascorbic acid (AA) otherwise known as VC. AA can restore TET activity via the facilitation of the Fe (III)/Fe (II) redox reaction. It has been implicated in the prevention and therapy of myeloid neoplasia (MN), caused by loss-of-function TET2 mutations (TET2MT). Mechanistically, the AA-based pharmacologic modulation of acetyltransferases and histone deacetylases could regulate the TET-dependent N- and C-terminal lysine acetylation albeit further investigations are required to clarify. Thus, this discovery could potentially suggest AA alone or the combination with class I/II HDAC inhibitor, sirtuin activators to act as therapeutic approaches for TET2M-based leukemia [142]. TET2 could be recruited and demethylate the promoter region of MMP9 to activate the expression of MMP9. Furthermore, the impact on the proliferation, migration, and invasion of HTR-8/SVneo cells in trophoblasts caused by downregulation of Tet2 could be rescued by AA, an activator of TET2 activity. Altogether, this suggests that MMP9 downregulation conferred by TET2 deficiency contributes to the pathogenesis of preeclampsia [143].
SLC2A3 is involved in the pathways such as the metabolism of water-soluble vitamins and cofactors as well as the HIF1 alpha pathway. Due to TET deficiency in particular TET2 in leukemogenesis, VC-based restoration of TET activity has been beneficial to AML therapy. However, SLC2A3 downregulation in AML severely blocked the absorption of VC, suggesting SLC2A3 as a potential biomarker for the VC-based therapy for AML [144].
Like TET1, TET2 deficiency could cause chemoresistance in head and neck cancer, GC, and BC cells. Failure to maintain the methylation/demethylation balance results in aberrant silencing or activation of the genes involved in tumorigenesis/suppression and contributes to pathogenesis, leading to chemotherapeutic resistance. TET2 is required to make some chemotherapeutic drugs (such as doxorubicin) functional for curing some cancers such as head and neck cancers via its demethylation activity to regulate gene expression and the resultant cell proliferation/migration. As an example, in response to doxorubicin, the physical interaction of TET2 with promyelocytic leukemia (PML) leads to the recruitment of TET2 to PML-positive nuclear bodies, thereby reactivating the silenced suppressor genes and subsequently impairing cell proliferation/migration. Thus, TET2 and PML could be the two promising therapeutic targets evidenced by the fact that PML KO abolished DNA modifications in response to doxorubicin. Indeed, the levels of PML and TET2 were positively correlated with overall survival in head and neck cancer patients [145].
TET2 is involved in the modulation of cisplatin resistance of GC cells as well in ways that significantly downregulated TET2 in cisplatin resistance SGC7901/DDP cells relative to the non-resistant cells. Accordingly, TET2 overexpression dramatically inhibited the cisplatin tolerance. As a mechanism, TET2 regulated the levels of interleukin-6, thereby modulating the cisplatin resistance via the regulation of histone acetylation. Thus, this study may uncover a novel mechanism of drug resistance in GC cells, potentially translating to the clinical approach for eradicating the drug resistance [146].
TET2 deficiency is also associated with chemoresistance in breast cancer cells. Clinically, chemoresistance to the poly(ADP-ribose) polymerase (PARP) inhibitors (PARPis) is common for treatment of breast and ovarian tumors caused by BRCA deficiency. In KB2P1.21 mouse mammary tumor cells, repressed TET2 expression led to the resistance to multiple PARPi, downregulating 5-hmC levels and protecting stalled replication forks (RFs) in the BRCA2-deficiency cells. TET2 expression-mediated 5-hmC abundance in the BRCA2-deficient mouse and human cancer cells induced the degradation of the stalled RFs via the recruitment of the base excision repair-associated apurinic/apyrimidinic endonuclease APE1, independent of BRCA2 status. Loss of 5-hmC and APE1 recruitment to stalled RFs due to TET2 deficiency promoted resistance to the chemotherapeutic cisplatin, suggesting a potential mechanism of the TET2-mediated epigenetic mark 5-hmC in resistance to PARPi and cisplatin by maintaining the integrity of stalled RFs [147].
TET2 was phosphorylated at tyrosines 1939 and 1964, which was activated by the cytokine-stimulated JAK2, leading to the enhanced interaction between TET2 and KLF1, an erythroid transcription factor. Thus, extracellular signals were linked to the epigenetic modifications mediated by TET2, leading to the upregulation of several oncogenes [148]. Sometimes, transcriptional activators that are unable to bind to DNA due to the lack of specific DNA binding domains can still recruit TET2 and activate specific target transcription factors via the ternary interaction among transcriptional coactivators. Via mammalian two-hybrid screening, multiple transcriptional regulators have been identified to interact with TET2. Of the identified TET2 interacting transcriptional activators, SNIP1, while possessing no specific DNA binding abilities, could recruit TET2 to several transcription factors, such as c-MYC. Thus, SNIP1 bridges TET2 to the promoters of c-MYC target genes via TET2-SNIP1-c-MYC ternary interactions to regulate target genes of these transcription factors [149].
Previous studies have indicated that TET2 functions as a tumor promoter via the aberrant AID pathway. In diffuse large B-cell lymphoma (DLBCL) patients who carry abnormal activation-induced cytidine deaminase (AID) expression, the enhanced demethylation mediated by TET2 on some proto-oncogene promoters could initiate or enhance oncogenesis. In this context, TET2 could function as an accomplice for DLBCL. Essentially, AID recruits TET2 to the promoter region of the dramatically expressed proto-oncogene Fanconi anemia complementation group A gene (FANCA), whereby the FANCA is activated via TET-catalyzed demethylation. Thus, AID, TET2, and FANCA work together to contribute to oncogenesis, suggesting that AID and TET2 could potentially become new therapeutic strategies for DLBCL [150].
Inactivating mutations of Tet2 are frequently found in human malignancies, suggesting the essential role of Tet2 in cellular transformation. Similar to AID in function of TET2, both MBD3 and MBD3L2 could specifically enhance the hydroxymethylation/demethylation activity of TET2 only, but not TET1 or TET3. Moreover, MBD3L2 could be functional more effectively than MBD3 via the enhancement of the binding affinity between the enzyme and the substrate of the methylated DNA. ChIP analysis identified the genomic binding sites co-occupied by MBD3L2 and TET2. The majority of the co-occupied regions belong to promoter elements for the genes involved in tumorigenesis such as XRCC6 and LDHB or the genes relating to metabolic regulation. Since TET2 does not contain the CXXC domain, it could be recruited by MBD3 and MBD3L2 or possibly by all the MBD family members to the appropriate genomic regions for the downstream events [151].
Significant attention has been paid to the functions of TET1 and TET2 in human cancers, particularly TET1and its role in oncogenesis or anti-tumor activity. Relative to TET1 and TET2, there remains limited information regarding the functions of TET3 in human cancers. However, based on the current studies, TET3 similarly functions as a tumor suppressor and promoter in a cancer-type-dependent manner.
TET3 has been proposed to be a candidate tumor suppressor for certain cancers such as GC, colorectal cancers (CRC), glioblastoma, and neck squamous cell carcinoma (HNSCC). Shift mutation of Tet3 gene harboring the mononucleotide repeats in the coding region leads to loss of TET3 expression in GC and CRC [152]. TET3 functioned as a tumor suppressor for glioblastoma tumorigenesis via the alteration of the 5-hmC landscape, evidenced by the fact that epigenetic downregulation of TET3 significantly declined the genome-wide 5hmC levels and promoted glioblastoma tumorigenesis, and vice versa. This suggests TET3 as a potential therapeutic target for glioblastoma [153].
To understand the functions of TET3 in tumorigenesis and the risk of disease recurrence in HNSCC, the methylation profiles of Tet1, Tet2, and Tet3 genes were generated in more than 200 tumor samples from HNSCC patients. It turned out that the simultaneous methylation of Tet genes was correlated with reduced disease-free survival and methylation levels of the tumor suppressor genes. In particular, the methylation events mediated by TET3 were independently and positively correlated with aggressive tumor performance and DNA methylation status globally in HNSCC, suggesting the predominant functions of TET3 over TET1 and TET2 in this regard [154].
Like TET2, TET3 serves as cancer promoter via the maintenance of cancer stem
cells (CSCs) such as glioma stem cells (GSCs). Activation of the integrin
TET3 also functions as a cancer promoter by maintaining the stemness of some cancer cells such as in esophageal squamous cell cancer (ESCC). Significant accumulation of lipopolysaccharide (LPS) was detected in ESCC, facilitating stemness of the ESCC cells via the sequential stimulation of the TET3/p38/ERK-MAPK pathway in ESCC and consequently leading to poor ESCC patient survival. Enhanced TET3 expression increased the expression of its target HOXB2 via the elevation of 5-hmC levels at the HOXB2 promoter. ChIP results demonstrated that HOXB2 bound to the genomic loci of Nanog and c-Myc initiated or facilitated the stemness of ESCC. Therefore, LPS served as an ESCC promotor via ESCC stemness through the activation of the LPS-TET3-HOXB2 signaling axis potentially serving as a novel therapeutic target for ESCC [156].
Some environmental toxicants, such as glyphosate, could render TET3 functions as tumor promoter as well. The known effect of glyphosate on estrogen-regulated signaling leads to its suspected pathogenesis to breast cancer. However, there is no evidence to show that the nonneoplastic MCF10A cells treated with the glyphosate alone could induce tumorigenesis. This suggests a synergistic effect of some environmental toxicants such as glyphosate with other endogenous factors triggering tumorigenesis. Unlike the UP peptide, a strong demethylating agent and a cancer promoter function via the DNMT1/PCNA/UHRF1 pathway. Glyphosate significantly elevated demethylation levels by enhancing TET3 activity. Synergistic exposure to glyphosate plus upregulation of miR182-5p significantly led to tumor development. Consistently, downregulation of miR182-5p or treatment with dimethyloxallyl glycine, an inhibitor of the TET pathway, could prevent oncogenesis. Thus, this discovery indicated that the low pressure from the TET pathway-mediated DNA hypomethylation could stimulate the cells for tumorigenic responses if in the presence of another potential risk factor [157].
Recurrent TET2 loss-of-function mutation events frequently occur, while TET3 mutations are infrequent in acute myeloid leukemia (AML) and in preleukemic hematopoietic stem cells (PHSCs). However, TET3 expression in HSCs was gradually downregulated during aging. Mouse models bearing the various degree of TET deficiency have been employed to test the relative contribution of TET2 or TET3 deficiency to AML pathogenesis. It demonstrated that simultaneous loss of function of TET2 and TET3 led to AML with the shortest survival latency, while the AML from a single TET loss of function could survive longer. Spontaneous inactivation of residual Tet2 or Tet3 alleles has been implicated as a recurrent genetic event during the development of AML owing to TET insufficiency [158].
Previous studies have concluded that activation of both TET2 and TET3 could
enhance tumor suppression. In the genome of AML, the two mutually
exclusive mutations recurrently take place, including the mutations of the
heterozygous inactivating TET2 and IDH1/2. Phenotypically, VC treatment repressed
cell proliferation and enhanced leukocyte differentiation by the enhanced
expression and activation of TET2 through VC. Epigenomically, TET2 was recruited
by master transcription factors such as CEBP
VC could significantly enhance the expression of both TET2 and TET3 but not TET1. VC-mediated 5-hmC gains and enhanced demethylation lead to the alteration of the 5-hmC landscape in the epigenome. However, this study failed to uncover the mechanism of how the expression of the genes involved in the proliferation of leukocytes is repressed, paradoxical to TET2-conferred demethylation and activation [159].
Interplay of TET members with DNMTs renders TET function as tumor suppressors. The coordinate interplay of TETs and DNMTs regulates the balance of methylation/demethylation in accordance with the developmental stage and physiological conditions in response to environmental exposure or disease pathogens. In addition to methylation and demethylation enzymes, the components such as ncRNAs (miRNAs, lncRNAs, circRNAs) and other key regulators for tumorigenesis are involved in the interplay as well.
Significant downregulation of TET1 and TET2 and dramatic upregulation of DNMT1 in papillary thyroid carcinoma (PTC) were associated with elevated levels of 5-mC and reduced levels of 5-hmC. Hypermethylation was detected in the Tet2 promoter region in the known thyroid carcinoma patients. The hypermethylation of TET1 and TET2 could serve as biomarkers for thyroid oncogenesis. In addition, suppressed Tet1 and Tet2 in K1-cells could be reactivated by treating the K1-cells with combined demethylating agents L-AA and 5-AzaC, suggesting that both TET1/2 and DNMT1 could be potential therapeutic targets for thyroid cancer [160].
Intrinsic and external environmental factors may contribute genetically and epigenetically to the pathogenesis of clonal hematopoiesis of indeterminate potential (CHIP), shrinking the diversity of the blood pool and increasing the risk for leukemia and cardiovascular disease in the elderly. Genetically, mutations in leukemia driver genes, such as Jak22, Tet2 Asxl1, and Dnmt3A, have been implicated in the generation of CHIP. Some environmental factors such as chemokines and cytokines have been epigenetically associated with CHIP. However, the genetic and epigenetic basis of CHIP onset, progression, and subsequent disease propagation remains to be investigated [161].
It has been shown that the interplay of TET3/DNMT3B mRNAs as novel competing endogenous RNAs (ceRNAs) functions as a suppressor through PTEN inactivation. PTEN inactivation caused by multiple genetic and epigenetic regulatory mechanisms contribute to oncogenesis. Epigenetically, ceRNAs in ncRNA dependent and coding independent regulation of the genes have been implicated in the regulation of PTEN expression. mRNAs of both Dnmt3B and Tet3 collectively function as novel ceRNAs of PTEN on the basis of sharing multiple miRNAs targeting their 3’UTRs, potentially affecting their dose sensitive tumor suppressive function. Reciprocal 3’UTR overexpression leads to competitive miRNA sequestration and subsequent activation of the PTEN pathway. Indeed, overexpression of 3’UTR for Dnmt3B or Tet3 strongly enhanced apoptosis and attenuated cell migration and evasion. Therefore, this study suggests that the dictatorship conferred by the number of miRNAs they share, and potentially by the existence of other ceRNA networks in which they participate, could determine the orchestrated phenotype [162].
The interplay of Dnmt1, Dnmt3A, Tet1,
Tet3, Jmjd3, and Utx could induce the conversion of cancer
cells to the senescent state. Cellular senescence serves as a tumor-suppressive
mechanism via the blocking of cell proliferation in response to stress. However,
the senescent tumor cells can re-enter the cell cycle to become cancer stem
cells, leading to relapse after chemotherapy. While the human embryonic
fibroblasts MRC5 and WI-38 cultured in stem cell medium (SCM) became senescent,
the cancer cells A549 and 293T cultured in SCM were converted to cancer stem
cells, suggesting different reprogramming performed in a culture
environment-dependent manner. Molecularly, SCM-induced senescence was associated
with the upregulation of genes for growth arrest. The overexpression of
Dnmt1, Dnmt3A, Tet1, Tet3, Jmjd3,
and ubiquitously transcribed tetratricopeptide repeat X chromosome (Utx) proteins
could activate senescence-associated beta-galactosidase (SA-
Interplay of TETs and miRNAs could regulate oncogenesis. As one of the
pathological mechanisms for myelofibrosis (MF) resistance to JAK2V617F inhibitor
therapy, miR-543 was found to be significantly upregulated and associated with
downregulation of TET1 and TET2 in the chemoresistance cells both in patients and
in vitro. Therefore, miR-543 may serve as a potential biomarker for the
prognosis of MF patients with a high risk of chemotherapy resistance.
Additionally, this potential biomarker could become a therapeutic target for MF
[164]. Similarly, chronic inflammation causes downregulation of
TET expression by upregulation of miRNAs such as miR20A, miR26B, and miR29C that
target Tets, likely via the activation of NF-
Interplay of TETs and key factors such as LGR5 and BMI1 involved in tumorigenesis could contribute to cancer promotion. Expression upregulation of LGR5, BMI1, TET1, and TET2 was detected in CD133 and EpCAM positive Cancer Stem-like Cells (CSCs), although there was no significant difference at the transcriptional level of TET3. Thus, high-level expression of the four genes may become a reliable criterion for characterization of the cells in colon cancer for the sake of developing specific targets for therapy of colon cancer [166].
Functional alteration of DNMTs and TETs has been confirmed in a variety of cancer types. One of the mechanisms refers to oncogene Myc-based deregulation of TET1 and TET2 expression. In T-cell acute lymphoblastic leukemia (T-ALL), overexpression of TET1 and suppression of TET2 were detected in a MYC-dependent manner, leading to the 5-mC/5-hmC patterns associated with tumor cell-specific gene expression. KD of Tet1 or overexpression of Tet2 in T-ALL leads to epigenome-wide alterations in 5-mC and 5-hmC landscape patterns, abolishing the MYC-conferred phenotype via inhibition of cell proliferation. Further studies displayed that TET1 enhanced the expression of the genes involved in ribosomal biogenesis and translational control. This suggests that the coordinated interplay between the components of the DNA (de)methylating machinery contributed to MYC-driven tumor maintenance, highlighting the potential application of specific TET enzymes for therapeutic strategies [167].
Interplay of TET2 and TET3 could enhance tumorigenesis. Significant upregulation of TET expression, particularly TET2 and TET3, is associated with high levels of 5-hmC, enhancing the cell proliferation in adenomas. Gradual elevation of UHRF1 expression was detected, and UHRF2 was positively correlated with 5-hmC levels in higher proliferative adenoma samples. This discovery indicated that epigenetic markers 5-hmC/5-mC could functionally contribute to the pathogenesis of pituitary neuroendocrine tumors (PitNET) via altering the epigenome in adenoma cells. Thus, 5-hmC/5-mC could serve as potential biomarkers as well as therapeutic targets for PitNET [168].
Interplay of TET members and -microbiota-colorectal could function as cancer suppressors as well. Whole-genome bisulfite sequencing has characterized the TET 2/3-dependent epigenetic alteration at regulatory elements by exposure to the intestinal microbiota. This epigenetic alteration activated a subset of ‘early sentinel’ response genes to maintain intestinal homeostasis via the regulation of profound DNA methylation and chromatin accessibility at regulatory elements. This led to upregulated expression of the genes that orchestrate in colitis- and colon-cancer-associated functions [169]. The mechanism of how the epigenetic reprogramming was altered by the intestinal microbiota may contribute to our understanding to the pathogenesis of colorectal tumorigenesis. More importantly, TET2/3 may potentially become therapeutic targets or prevent colorectal cancer caused by inflammatory bowel disease [170].
Dramatic breakthroughs in high throughput sequencing technologies significantly speed up the progress in epigenetic study. The main players implicated in epigenetic regulation, such as TETs, DNMTs, 5-mC, and 5-hmC, have received significant attention genetically, epigenomically, biochemically, and molecularly. The development of Tet-assisted bisulfite sequencing (TAB-Seq) [171] has enabled us to track the distribution of 5-hmC in the whole genome at a single-base resolution level. More recently, new techniques in TET-assisted pyridine borane sequencing (TAPS-seq) and Bisulfite-free direct detection of 5-mC and 5-mC [172] facilitate to track the distribution of 5-mC and 5-hmC in the whole genome at a single-base resolution level simultaneously. TAB-seq, TAPS-seq, single RNA-seq, and proteomics have greatly enhanced our detection of the precise and dynamic distribution of the essential epigenetic markers alongside their contribution to the regulation of gene expression. These advances in methodology efficiently facilitate the investigation on the contribution of the writers, erasers, and readers of 5-mC and 5-hmC to the pathogenesis of disease such as neurological disorders and cancers.
However, many unknown molecular and epigenetic players that contribute to the pathogenesis of cancer and other human diseases remain under investigation. Currently, the individual members of writers and erasers for 5-mC have been identified and characterized molecularly. Some readers specific for 5-mC and 5-hmC were identified as well. However, the present functional study on the TET proteins is simply focused on demethylation/hydroxymethylation-based regulation of the 5-hmC landscape in the epigenome, the subsequent up-or-down-regulation of gene expression, and the identification of tumor suppressors or promoters via the known mechanisms. Therefore, the networks that cover their coordination with other regulatory factors in orchestrating the landscape of the epigenome in response to the developmental stage, reproduction, environmental stresses, and tumorigenesis are still at the infancy stage.
Given that only a small percentage of 5-hmC is destined to serve as an intermediate for demethylation, the large portion of 5-hmC functions as “decoration” of the landscape in the epigenome. Therefore, the mechanism of how the epigenetic information stored in the landscape is transformed into a biological effect will be critical for further understanding the epigenetic regulation of development, reproduction, tumor initiation, and progression. Moreover, in terms of the functions of TET proteins, we may simply understand the catalytic role in oxidation of 5-mC and 5-hmC. ogically, such huge proteins may contain multiple functional domains in addition to catalytic domains, each of which the unknown domains potentially have its specific function to be identified, such as interactions with regulatory factors involved in essential signaling pathways. Future studies on the unknown domains and the corresponding functions will provide important information for dissection of the pathogenesis.
Formerly, only a limited number of readers for both 5-mC and 5-hmC have been identified. Therefore, more efforts are required to identify and characterize more readers/repellents and the related interaction factors, helping us further understand how and where the TETs are recruited to shape the dynamic 5-hmC/5-mC landscape. It is of importance to identify more factors that physically interact with TET members. To this end, CRISPR and organoid tools could provide important support via genome editing and human organoid-based modeling of human cancers as well as other diseases. In addition, chemical biology approaches could be employed for drug screening to identify small molecule drugs that target the 5-mC/5-hmC machinery or the signaling pathways. More importantly, the identified readers, repellants, TET interacting factors, and small molecule drugs could become promising therapeutic targets for cancers and other human diseases.
CM, HS, SX, and YL wrote and revised manuscript, YLIU and XY revised manuscript and prepared some figures.
Not applicable.
Thanks to all the peer reviewers for their opinions and suggestions.
Financially supported by Shandong Provincial Natural Science Foundation China (to SLX) # ZR2015HM024, and # 2019GSF108066; IIFDU and SFR for ROCS, SEM).
The authors declare no conflict of interest.