2. Introduction
Regulation of tissue factor (TF) activity and its release as microvesicles is
imperative to ensure adequate coagulation during injury, without endangering the
precipitation of thrombosis. Consequently, the procoagulant activity of TF is
precisely regulated through various mechanisms that influence the TF protein
[1, 2, 3]. Recently it was demonstrated that the activity and release of TF can be
controlled through the action of peptidyl-prolyl trans/cis isomerase 1
(Pin1) [4, 5]. Pin1 is a regulator of post-phosphorylation processes and binds to
the phosphoserine-proline (termed an MPM-2) motif [6, 7, 8, 9, 10, 11, 12, 13, 14, 15]. Normally
transcis isomerisation of the peptide bonds
occurs at a slow rate and the energy barrier for isomerisation is relatively
high. However, the rotation of the peptide bond between the phosphoserine and the
proline may be facilitated by enzymes including Pin1. The phosphorylation of
serine 258 within the cytoplasmic domain of TF produces such a
phosphoserine-proline motif which is a target for Pin1 [16, 17, 18]. Since the
trans configuration is the more prevalent of the two isomers, Pin1
preferentially catalyses the formation of the cis isomer. The
cis isomer is often not accessible to enzymes which bring about
subsequent modifications including ubiquitination [10, 12] and de-phosphorylation
[19]. The action of Pin1 on the cytoplasmic domain of TF prevents the
de-phosphorylation of serine 253 within the cytoplasmic domain of TF. This
extends the release of TF within microvesicles by preventing TF ubiquitination
[4, 20]. Pin1 has also been reported to prolong the activity of TF and also induce
the de novo expression of TF mRNA [5]. In addition to the procoagulant
activity, TF is known to promote signalling mechanisms that can give rise to cell
proliferation [21, 22, 23] or alternatively cell apoptosis [24, 25, 26]. The inability of
cells to dispose of excess TF efficiently appears to contribute to the induction
of cell apoptosis, mediated via p53 nuclear localisation and the expression of
Bax protein [24, 27, 28]. Moreover, although it has been established that Pin1 is
capable of influencing p53 function directly [13, 29, 30, 31, 32], there also appears to be
an independent regulation mediated through TF.
Previously, we examined the modifications and release of TF in the presence of
the non-selective Pin1 inhibitor, juglone. Recently, a number of small molecule
Pin1 inhibitors have been designed and tested for their effectiveness as a means
of regulating the cell function and particularly, in controlling the
proliferation of cancer cells [33, 34]. Using a combination of NMR based fragment
screening, co-crystallisation studies and structure-guided design, Potter
et al. [35, 36] reported the structure of several potent Pin1 inhibitors.
Initial studies using NMR screening identified the substance as 5-methylindole
2-carboxylic acid (1) (Fig. 1A) and from subsequent fragment
co-crystallisation studies, 5-methylbenzimidazole 2-carboxylic acid (2)
was identified as a more potent building block. Finally, this compound was
redesigned into the complex derivative 3 which had a Pin1 IC of
0.13 M. However, this proved ultimately to be of limited potential
since it lacked any cellular potency. To reduce the polar surface area of the
molecule, the benzimidazole was replaced with the 2-naphthyl group, using the
commercially available ()-2-naphthylalanine, to provide the derivative
2’-methyl-5’-(p-methoxyphenyl)-3’-furoyl-3-(2-naphthyl)--alanine
(4a). This compound proved to be less potent than 2 against Pin1
(IC 2.6 M) but was active in intact cells. In this study, we based
a set of small molecules on 5-(p-methoxyphenyl)-2-methylfuran-3-carbonyl
amide and modified the ‘head groups’ to modulate the inhibitor-protein
interactions. We synthesised and tested four compounds that differed in the amino
termini to contain -tryptophan (4b), -phenylalanine
(4c), and -tyrosine (4d), as well as the previously
reported 3-(2-naphthyl)--alanine (4a). We then examined the
effectiveness of these compounds to alter the activity, expression and release of
TF, as well as the potential to block Pin1 activity and its interaction with the
cytoplasmic peptide of TF. Finally, the influence of the compounds on the
promotion of cell apoptosis, p53 nuclear localisation and Bax/bax expression was
assessed.
 Fig. 1.
Fig. 1.
Structure (A) and synthesis (B) of potential Pin1 inhibitors. Synthesis
of the compounds is detailed in the supplementary material. The procedure for the
preparation of derivatives was modified substituting the
()-naphthylalanine moiety with other aromatic ()-amino acids;
tyrosine, tryptophan and phenylalanine. As a result, a set of four compounds were
prepared based on 5-(p-methoxyphenyl)-2-methylfuran-3-carbonyl amide and
synthesised to include (4a) 3-(2-naphthyl)--alanine,
(4b) -tryptophan, (4c) -phenylalanine and
(4d) -tyrosine, as the head-groups.
3. Material and methods
3.1 Synthesis and analysis of potential Pin1 inhibitor compounds
Following the identification of 4a as a functional compound,
derivatives in which the ()-naphthylalanine moiety was replaced with
other aromatic ()-amino acids (tryptophan, phenylalanine and tyrosine)
were envisaged. As a result, a set of four compounds (Fig. 1B) were synthesized
(see Supplementary material) based on
5-(p-methoxyphenyl)-2-methylfuran-3-carbonyl amide, with a view to
examining their structure-activity relationship.
Prior to testing of the biological potential of the synthesised compounds in
cells, the structure and purity of the compounds were confirmed. Purification was
achieved via column chromatography using Merck 200-300 mesh silica gel, and for
thin layer chromatography (TLC) Merck 60 mesh size pre-coated aluminium plates
were used. Visualization of TLC bands was achieved using a UV lamp at 254 nm. NMR
spectra were obtained using a Jeol JNM ECP400 spectrometer (Welwyn Garden City,
UK). ES-MS data were collected on HCT ultra ETD II mass spectrometer (KRSS Europe
BV, Veenendaal, Netherlands) and melting points were recorded using a
Fisher-Johns apparatus in open capillary tubes. CHN combustion elemental
microanalyses were performed using a Carlo-Erba EA1108 CHN Analyzer (Fisons,
Loughborough, UK).
3.2 Cell culture, determination of cell numbers and apoptosis
assays
MDA-MB-231 breast cancer cell lines (ATCC, Teddington, UK) were cultured in DMEM
containing 10% (v/v) FCS. Human dermal blood microvascular endothelial cells
(HDBEC; PromoCell, Heidelberg, Germany), devoid of endogenous TF were cultured in
MV media containing 5% (v/v) foetal calf serum (FCS) and growth supplements
(PromoCell). Cells (5 10) were seeded out into 48-well plates
and treated with the set of inhibitor compounds (100 M) or the DMSO
vehicle for 24 h. Cell numbers were determined by staining with crystal violet as
previously described [37, 38] and calculated from a standard curve. In addition,
cellular apoptosis was quantified using the TiterTACS™
Colorimetric Apoptosis Detection Kit (AMS Biotechnology, Abingdon, UK) according
to the manufacturer’s instructions [26, 39].
3.3 Pin1 activity assay
We previously showed that active Pin1 was capable of binding a synthetic
phosphorylated substrate peptide corresponding to the last 18 amino acids of the
cytoplasmic domain of TF [4]. A biotinylated form of the phosphorylated peptide
(biotin-RKAGVGQSWKENpSPLNVS) was synthesised (Biomatik, Ontario, Canada) and used
here as a means of detecting Pin1 binding activity in vitro. An additional
scrambled peptide (biotin-SWGNVSKLSAPRQGVNKE) was also included alongside as
control. The measurements were carried out as outlined previously [4]. Briefly,
the peptides (5 M final concentration) were diluted to 100
L with PBS and distributed (50 L per well) in a
NeutrAvidin-coated 96-well plate (Thermo Scientific, Warrington, UK) and
incubated for 2 h at room temperature to allow binding. The wells were washed
four times, each time with 300 L of PBST. Sets of wells were
supplemented with a range of synthetic inhibitor compounds (0–100
M) or DMSO vehicle. HRP-conjugated recombinant Pin1 protein was
then diluted 1:500 (v/v) in PBST, added to the wells (100 L) and
incubated for 1 h at room temperature. The wells were then washed a further four
times and developed with TMB One Solution (100 L; Promega
Corporation, Southampton, UK). Once the colour was developed the reactions were
stopped by the addition of 2M sulphuric acid (50 L) and absorptions
measured at 450 nm using a plate-reader. The concentrations of Pin1 were
determined from a standard curve made using HRP-conjugated recombinant Pin1
protein.
The isomerase activity of Pin1 was also examined using a previously described
procedure [4, 40]. Pin1 was incubated with penta-peptides,
Succ-ENSPL-pNitroanilide and Succ-ENpSPL-pNitroanilide and alterations in
spectroscopic absorption of the solution analysed. Briefly, samples (100
L) of the substrate peptides (0.5 M) were in turn
placed in a microcuvette. Recombinant HRP-conjugated Pin1 (10 nM final
concentration) was then pre-incubated (5 min) with the inhibitors and then added
to the cuvettes. The change in absorption at 315 nm over time was then
immediately monitored.
3.4 Cell-based factor Xa-generation assay
Cell surface TF-fVIIa activity was measured by modification of previously
described procedures [4]. MDA-MB-231 cells (5 10) were incubated
with the compound inhibitors (100 M) diluted in the reaction buffer
(100 L; HEPES-buffered saline (HBS) pH 7.4, containing 1% (w/v)
bovine serum albumin (BSA) and 5 mM CaCl) for 60 min. The cells were washed
and incubated with fVIIa (20 nM; Enzyme Research Labs, Swansea, UK) in the
reaction buffer for an additional 10 min and then supplemented with fX (100 nM),
together with fXa substrate (0.2 mM; Hyphen) diluted in the same buffer (100
L). The samples were incubated for 60 min to develop the colour.
Aliquots (150 L) were then transferred to a 96-well plate
containing 2% (v/v) acetic acid (50 L) and the absorptions
measured immediately at 405 nm. The amount of fXa generated was determined using
a standard curve prepared using fXa (Enzyme Research Labs, Swansea, UK). To
confirm the cell surface TF activity, cells were pre-incubated with the
inhibitory anti-TF antibody HTF1 (40 g/mL; eBioscience/Thermo
Scientific, Warrington, UK) prior to the addition of fVIIa.
3.5 Analysis of cell surface and microvesicles-associated TF
antigens
MDA-MB-231 cells (5 10) were seeded out into 48-well plates and
incubated with the inhibitor compounds as above (100 M) for 18 h.
The cells were then washed with PBS and fixed with 3% (v/v) formaldehyde. The
cells were incubated with an HRP-conjugated sheep anti-human TF antibody (100
L, Enzyme Research Labs., Swansea, UK) diluted 1:1000 (v/v) in PBS
for 1 h. The cells were then washed four times with PBS and developed using TMB
One Solution substrate Solution (100 L). Once the colour was
developed the reactions were stopped by the addition of 2M sulphuric acid (50
L) and absorptions measured at 450 nm using a plate-reader.
Microvesicle-associated TF antigen was measured using the Quantikine TF-ELISA kit
(R&D Systems, Abingdon, UK) according to the manufacturer’s instructions.
3.6 Western blot analysis of p53 and Bax proteins
Cells were incubated overnight with the compound inhibitors as above. The cells
were then lysed in Laemmli’s buffer containing a protease inhibitor cocktail
(Sigma Chemical Company, Poole, UK) and equal amounts were separated by 12%
(w/v) SDS-PAGE. The protein bands were transferred onto nitrocellulose membranes
and blocked with TBST (10 mM Tris-HCl pH 7.4, 150 mM NaCl, 0.05% Tween-20). The
membranes were then probed with a polyclonal rabbit anti-human p53 antibody (Cell
Signalling Technologies/New England Biolabs, Hitchin, UK) or a mouse monoclonal
anti-human Bax antibody (202; Santa Cruz Biotechnology, Heidelberg, Germany),
diluted 1:3000 (v/v) in TBST. The membranes were then washed with TBST and probed
with a goat anti-rabbit or goat anti-mouse alkaline phosphatase-conjugated
antibody (Santa Cruz Biotechnology, Heidelberg, Germany) diluted 1:1000 (v/v) and
incubated for 90 min. The bands were then visualised using the Western Blue
stabilised alkaline phosphatase-substrate (Promega Corporation Ltd, Southampton,
UK) and recorded. All quantifications were normalised against GAPDH which was
detected using a polyclonal goat anti-GAPDH antibody diluted 1:5000 (v/v) and
then detected using an alkaline phosphatase-conjugated donkey anti-goat-IgG
antibody (Santa Cruz Biotechnology, Heidelberg, Germany) diluted 1:2000 (v/v).
3.7 Quantification of TF and bax mRNA expression by quantitative
real-time RT-PCR
Total RNA was isolated using the TRI-reagent system (Sigma Chemical Company,
Poole, UK) from 2 10 cells and 100 ng of total RNA was used for
each reaction. The relative amounts of TF or bax mRNA were determined using
QuantiTect primer sets to detect either TF or bax, in conjunction with
-actin (Qiagen, Manchester, UK). The reaction was carried out at an
annealing temperature of 60 C for 1 min using the
GoTaq® 1-Step RT-qPCR System (Promega Corporation Ltd,
Southampton, UK) on an iCycler thermal cycler (Bio-Rad, Hemel Hempstead, UK) for
40 cycles. Following amplification, the amounts of mRNA were determined using the
2 method and ratios were calculated [41].
3.8 Analysis of p53 nuclear localisation by fluorescence microscopy
MDA-MB-231 cells (10) were seeded out into 35-mm glass-bottom with 10 mm
-well dishes and incubated for 4 h at 37 C. The medium
was aspirated and replaced with 100 L of medium supplemented with test
reagents (100 M). Sets of cells were treated with of TNF-
(10 ng/mL) or DMSO and were used as positive and negative controls, respectively.
The media were removed after 18 h and the cells were washed twice with PBS (200
L) and fixed using 4% (v/v) paraformaldehyde. After three further washes
with PBS, the cells were permeabilised with 0.2% (v/v) Triton X-100 diluted in
PBS and incubated at room temperature for a further 10 min. The samples were then
blocked for 1 h with PBS containing 3% (w/v) bovine serum albumin (BSA). The
cells were then washed a further three times and probed with a rabbit polyclonal
anti-human p53 antibody diluted 1:250 (v/v) in PBS/BSA buffer (100
L) and incubated overnight, at 4 C. After a further three
washes with PBS, the samples were incubated for 1 h with a Northern Lights-637
donkey anti-rabbit antibody (R&D Systems, Abingdon, UK) diluted 1:100 (v/v) in
PBS/BSA buffer (100 L), in the dark. The cells were then washed
another two times with PBS and stained with DAPI (2 g/mL). Images
were acquired using a Zeiss Axio Vert.A1 inverted fluorescence microscope with a
40 magnification (Carl Zeiss Ltd, Welwyn Garden City, UK). The
localisation of p53 within the nuclei were determined using ImageJ program
(version 1.48v, LOCI, University of Wisconsin, Madison, WI, USA), in 10 fields of
view from each assay and Mander’s coefficient determined [42, 43].
3.9 Approximation of binding of compounds to Pin1
The structures of the four compounds were constructed using the Alchemy program
(Tripos Associates Inc., St Louis, USA) and then saved in Brookhaven format
(PDB). The crystal structures of Pin1 (1PIN) was obtained from Brookhaven format
(PDB). The location and efficiency of binding of the compounds to Pin1 was
estimated using Autodock 4v2.6 [44]. The Autodock graphical interface
AutoDockTools 1.5.6 was used, the polar hydrogens were retained and partial
charges added to the proteins using the Gasteiger charges. The search space was
limited to an area of 20 20 20 Å, centred around the
hydroxyl group of Ser18 in the enzymatic site of Pin1. For each enzyme, 25
ligand orientations (poses) were examined and ranked according to the
scoring-function and inhibition coefficient calculated.
3.10 Statistical analysis
All data represent the calculated mean values from the number of experiments
stated in each figure legend the calculated standard error of the mean.
Statistical analysis was carried out using the Statistical Package for the Social
Sciences v21 (SPSS Inc. Chicago, IL, USA). Significance was determined using
one-way ANOVA (analysis of variance) and Tukey’s honesty significance test or
where appropriate, by paired t-test.
4. Results
4.1 The influence of synthesised inhibitors on Pin1 binding and
activity
To determine the inhibitory potential of the synthesised compounds, the ability
of the compounds to prevent the substrate binding and isomerase activities of
Pin1 was examined. The enzyme used was recombinant HRP-conjugated Pin1 and the
target substrate peptide was biotin-RKAGVGQSWKENpSPLNVS (from the cytoplasmic
domain of TF) which was described and confirmed previously as a suitable target
for Pin1 binding [4]. Inclusion of -tryptophan (substance 4b)
and -tyrosine (substance 4d) head-groups inhibited the Pin1
binding to the substrate peptide with latter being the more efficient inhibitor
(Fig. 2A). In contrast, inclusion of -phenylalanine (substance
4c) head-group had no significant outcome on Pin1 binding while the
addition of 3-(2-naphthyl)--alanine (substance 4a) marginally
enhanced Pin1 binding. In addition to the binding assay, the isomerase activity
of Pin1 towards a pentapeptide (Succ-ENpSPL-pNitroanilide) was measured
spectroscopically in the presence of the synthesised inhibitor substances (Fig. 2B). Analyses of these samples showed a clear decrease in Pin1 isomerase activity
with the -tyrosine derivative (substance 4d) but not
phenylalanine (substance 4c) derivative. In contrast, an increase in
Pin1 isomerase activity was detected on inclusion of
3-(2-naphthyl)--alanine (substance 4a) and
-tryptophan (substance 4b) head-groups (Fig. 2B).
 Fig. 2.
Fig. 2.
The influence of the synthesised inhibitors on Pin1 binding and
activity. (A) A biotinylated form of the phosphorylated TF peptide
(biotin-RKAGVGQSWKENpSPLNVS) was used to capture Pin1 in vitro along
with a scrambled peptide (biotin-SWGNVSKLSAPRQGVNKE) as control. The peptides (5
M final concentration) were diluted to 100 L with PBS
and distributed (50 L per well) in a NeutrAvidin-coated 96-well and
incubated for 2 h at room temperature to allow binding. The wells were washed
four times, each time with 300 L of PBST. Sets of wells were
supplemented with a range of synthetic inhibitors (0–100 M) or
DMSO vehicle. HRP-conjugated recombinant Pin1 protein was diluted 1:500 (v/v) in
PBST, added to the wells (100 L) and incubated for 1 h at room
temperature. The wells were then washed a further four times and developed with
TMB One Solution (100 L). Once the colour was developed the
reactions were stopped by the addition of 2M sulphuric acid (50 L)
and absorptions measured at 450 nm using a plate reader. The concentrations of
Pin1 were determined from a standard curve made using HRP-conjugated recombinant
Pin1 protein (n = 3, * = p 0.05). (B) Pin1 was incubated with
penta-peptides, Succ-Glu-Asn-Ser-Pro-Leu-pNitroanilide and
Succ-Glu-Asn-phosphoSer-Pro-Leu-pNitroanilide and alterations in spectroscopic
absorption of the solution analysed. Briefly, samples (100 L) of
the substrate peptides (0.5 M) were in turn placed in a
microcuvette. Recombinant HRP-conjugated Pin1 (10 nM final concentration) was
then pre-incubated (5 min) with the inhibitors and then added to the cuvettes.
The change in absorption at 315 nm over time was then immediately monitored (n =
3, * = p 0.05).
4.2 The influence of synthesised inhibitors on TF activity,
cell-surface and microvesicle-associated TF antigen and TF mRNA expression
To assess the direct influence of the inhibitors on TF activity, MDA-MB-231
cells were pre-incubated for 60 min with the synthesised substances and TF
activity measured using a chromogenic fXa-generation assay. Inclusion of
-tryptophan (substance 4b) and -tyrosine (substance
4d) head-groups in the compounds inhibited the thrombin generation by
38% and 31% respectively (Fig. 3A). In contrast, reductions in TF activity, on
inclusion of 3-(2-naphthyl)--alanine (substance 4a) and
-phenylalanine (substance 4c) derivatives were not
significant. Therefore, only the alterations observed on with
-tryptophan (substance 4b) and -tyrosine (substance
4d) derivatives were assumed to be specific.
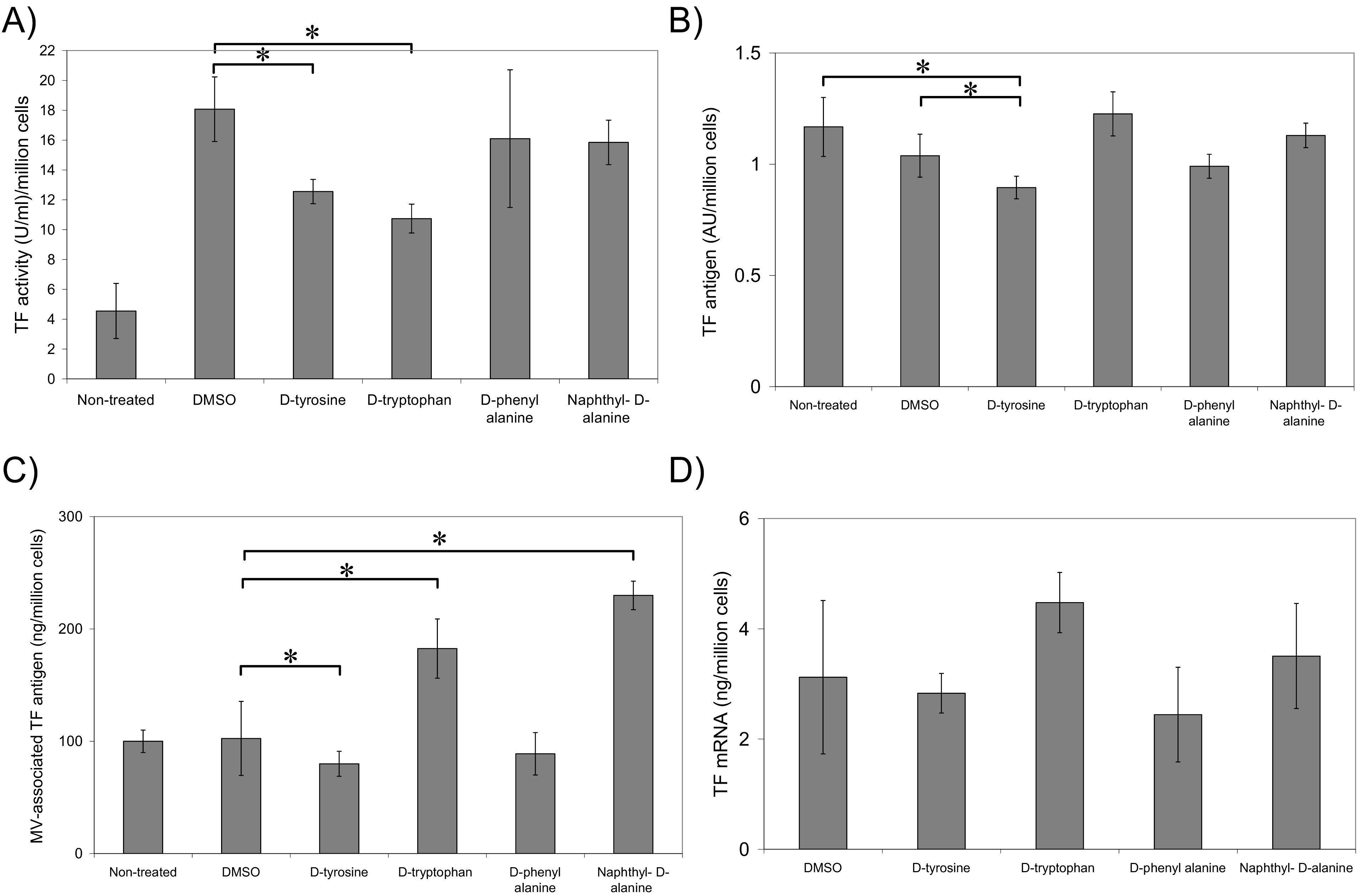 Fig. 3.
Fig. 3.
The influence of the synthesised inhibitors on TF activity,
antigen and mRNA levels. (A) MDA-MB-231 cells (5 10) were
incubated with the inhibitors (100 M) diluted in the reaction
buffer (100 L; HEPES-buffered saline (HBS) pH 7.4, containing 1%
(w/v) bovine serum albumin (BSA) and 5 mM CaCl) for 60 min. The cells were
washed and incubated with fVIIa (20 nM) in the reaction buffer for an additional
10 min and then supplemented with fX (100 nM), together with fXa substrate (0.2
mM) diluted in the same buffer (100 L). The samples were incubated
for 60 min to develop the colour. Aliquots (150 L) were then
transferred to a 96-well plate containing 2% (v/v) acetic acid (50
L) and the absorptions measured immediately at 405 nm. The amount
of fXa generated was determined using a standard curve prepared using fXa (n = 5,
* = p 0.05). (B) MDA-MB-231 cells (5 10) were
seeded out into 48-well plates and incubated with the inhibitors as above (100
M) for 18 h. The cells were then washed with PBS and fixed with 3%
(v/v) formaldehyde. The cells were then incubated with an HRP-conjugated sheep
anti-human TF antibody (100 L) diluted 1:1000 (v/v) in PBS for 1 h.
The cells were then washed four times with PBS and developed using TMB One
Solution substrate Solution (100 L). Once the colour was developed
the reactions were stopped by the addition of 2M sulphuric acid (50
L) and absorptions measured at 450 nm using a plate reader (n = 5,
* = p 0.05). (C) The TF antigen associated with the microvesicles
was measured using the Quantikine TF-ELISA kit according to the manufacturers’
instructions (n = 3, * = p 0.05). (D) Total RNA was isolated using
the TRI-reagent system from 2 10 cells and 100 ng of total RNA
was used for each reaction. The relative amounts of TF mRNA were determined using
QuantiTect primer sets to detect TF in conjunction with -actin. The
reaction was carried out at an annealing temperature of 60 C for 1 min
using the GoTaq® 1-Step RT-qPCR System on an iCycler thermal
cycler for 40 cycles (n = 3, * = p 0.05).
Incubation of MDA-MB-231 cells with compound 4d, containing
-tyrosine head-group, resulted in low but significant reduction in
cell-surface TF antigen (Fig. 3B) and TF release within microvesicles (Fig. 3C).
Moreover, treatment of cells with either substances containing
3-(2-naphthyl)--alanine (substance 4a) or
-tryptophan (substance 4b) head groups resulted in substantial
increases in the release of TF within cell-derived microvesicles (Fig. 3C).
Incubation of MDA-MB-231 cells for 6 h with any of the four synthesised
compounds, did not result in significant changes in TF mRNA expression (Fig. 3D).
4.3 The influence of synthesised inhibitors on cellular apoptosis
Incubation of MDA-MB-231 cells with substances containing the
3-(2-naphthyl)--alanine (substance 4a), -tryptophan
(substance 4b) and -tyrosine (substance 4d)
head-groups resulted in approximately 22%, 48% and 55% reduction in cell
numbers respectively (Fig. 4A) and were associated with significant increases in
DNA-fragmentation as measured by the end-labelling TUNEL assay (Fig. 4B). In
contrast, the inclusion of -phenylalanine (substance 4c) was
ineffective. Importantly, incubation of the HDBEC primary cells which are devoid
of TF, did not have any detectable influence on cell numbers of the rate of cell
apoptosis (Fig. 4C).
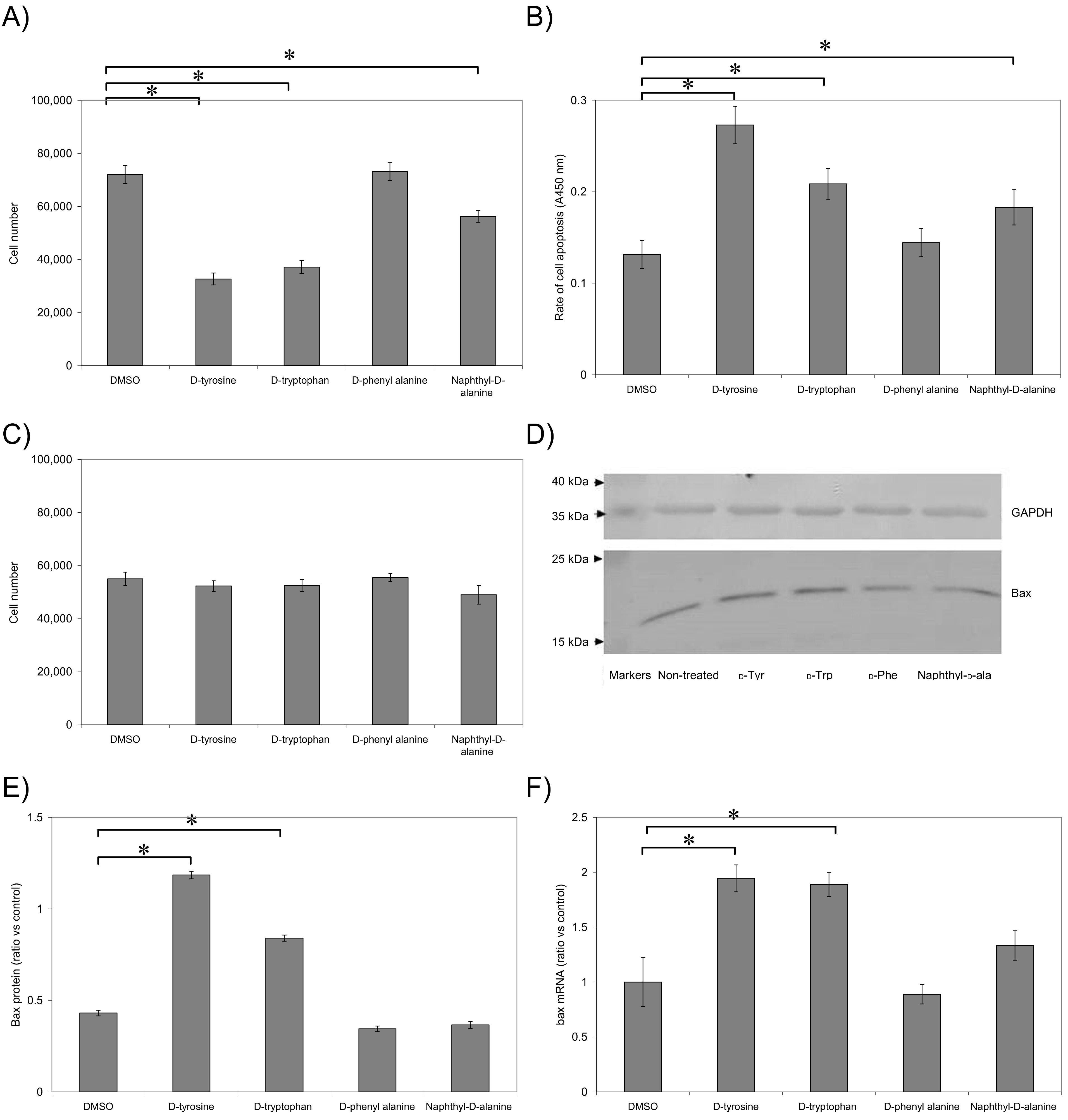 Fig. 4.
Fig. 4.
Examination of the pro-apoptotic potential of the synthesised
inhibitors. (A) MDA-MB-231 cells (5 10) were seeded out into
48-well plates and treated with the set of inhibitors or the DMSO vehicle. Cell
numbers were determined by staining with crystal violet and calculated from a
standard curve (n = 5, * = p 0.05). (B) Cellular apoptosis was
quantified using the TiterTACS™ Colorimetric Apoptosis Detection
Kit according to the manufacturer’s instructions (n = 3, * = p
0.05). (C) HDBEC (5 10) were seeded out into 48-well plates and
treated with the set of inhibitors or the DMSO vehicle. Cell numbers were
determined by staining with crystal violet and calculated from a standard curve.
(n = 3). (D) Cells (5 10) were treated as above and lysed in
Laemmeli’s buffer containing a protease inhibitor cocktail. Equal amounts were
separated by 12% (w/v) SDS-PAGE and the protein bands were transferred onto
nitrocellulose membranes and blocked with TBST. The membranes were then probed
with a mouse monoclonal anti-human Bax antibody (202), diluted 1:3000 (v/v) in
TBST. The membranes were washed with TBST and probed with a goat anti-rabbit or
goat anti-mouse alkaline phosphatase-conjugated antibody diluted 1:1000 (v/v) and
incubated for 90 min. The bands were then visualised using the Western Blue
stabilised alkaline phosphatase-substrate and recorded (Images are representative
of 3 separate experiments). (E) All quantifications were normalised against GAPDH
which was detected using a polyclonal goat anti-GAPDH antibody diluted 1:5000
(v/v) and then detected using an alkaline phosphatase-conjugated donkey
anti-goat-IgG antibody diluted 1:2000 (v/v) (n = 3, * = p 0.05). (F)
Total RNA was isolated using the TRI-reagent system from 2 10
cells and 100 ng of total RNA was used for each reaction. The relative amounts of
bax mRNA was determined using QuantiTect primer sets to detect bax in conjunction
with -actin. The reaction was carried out at an annealing temperature of
60 C for 1 min using the GoTaq® 1-Step RT-qPCR System
on an iCycler thermal cycler for 40 cycles (n = 3, * = p 0.05).
4.4 The influence of synthesised inhibitor compounds on Bax
expression
To further confirm the mechanism of apoptosis in MDA-MB-231 cells, following
treatment with the inhibitors the expression of Bax protein and bax mRNA were
measured. -tyrosine derivative (substance 4d) and to a lesser
extent -tryptophan derivative (substance 4b) resulted in
increased expression of both Bax protein (Fig. 4D,E) and bax mRNA (Fig. 4F)
while the 3-(2-naphthyl)--alanine (substance 4a) and
-phenylalanine (substance 4c) derivatives were ineffective.
4.5 The influence of synthesised inhibitor compounds on p53
expression and localisation
Finally, since Pin1 can influence cell apoptosis through altering the activity
and stability of p53 protein, the influence of synthesised inhibitor substances
on the expression and nuclear localisation of p53 was examined. Incubation of
MDA-MB-231 cells with -tyrosine (substance 4d) containing
compound resulted in increased nuclear localisation of p53 compared to the
control sample, but was not significant with compounds containing the
3-(2-naphthyl)--alanine (substance 4a), -tryptophan
(substance 4b) and -phenylalanine (substance 4c)
head-groups (Fig. 5A,B). Moreover, incubation of the cells with any of the four
compounds did not alter the amount of p53 proteins as measured by western blot
(Fig. 5C).
 Fig. 5.
Fig. 5.
The influence of the synthesised inhibitors on nuclear
localisation of p53. Cells were seeded out into 35-mm glass-bottom with 10 mm
-well dishes were seeded 10 MDA-MB-231 cells and incubated
for 4 h at 37 C in an incubator. The medium was aspirated and replaced
with 100 L of medium supplemented with test agents (100 M).
Sets of cells were treated with of TNF- (10 ng/mL) or used untreated
and used as positive and negative controls, respectively. The media were removed
after 18 h and the cells were washed twice with PBS (200 L) and fixed
using 4% (v/v) paraformaldehyde. After three washes with PBS, the cells were
permeabilised with 0.2% (v/v) Triton X-100 diluted in PBS and incubated at room
temperature for a further 10 min. The samples were then blocked for 1 h with PBS
containing 3% (w/v) bovine serum albumin (BSA). The cells were then washed a
further three times and probed with a rabbit polyclonal anti-human p53 antibody
diluted 1:250 (v/v) in PBS/BSA buffer (100 L) and incubated
overnight, at 4 C. After a further three washes with PBS, the samples
were incubated with a Northern Lights-637 donkey anti-rabbit antibody diluted
1:100 (v/v) PBS/BSA buffer (100 L), for 1 h in the dark. The cells
were washed another two times with PBS and stained with DAPI (2
g/mL). (A) Images were acquired using a Zeiss Axio Vert.A1 inverted
fluorescence microscope with a 40 magnification (Images are
representative of 10 field of view from 3 separate experiments). (B) The
localisation of p53 within the nuclei were determined using ImageJ, in 10 fields
of view from each assay and Mander’s coefficient determined (n = 3, * =
p 0.05). (C) Cells (5 10) were treated as above and
lysed in Laemmeli’s buffer containing a protease inhibitor cocktail. Equal
amounts were separated by 12% (w/v) SDS-PAGE and the protein bands were
transferred onto nitrocellulose membranes and blocked with TBST. The membranes
were then probed with a polyclonal rabbit anti-human p53 antibody. The membranes
were washed with TBST and probed with a goat anti-rabbit or goat anti-rabbit
alkaline phosphatase-conjugated antibody diluted 1:1000 (v/v) and incubated for
90 min. The bands were then visualised using the Western Blue stabilised alkaline
phosphatase-substrate and recorded. (Images are representative of 3 separate
experiments).
4.6 Approximation of binding of compounds to Pin1
The interactions of the four compounds with Pin1 enzyme were examined using the
crystal structure of Pin1 (1PIN). The location (Fig. 6) and efficiency of binding
of the four compounds to Pin1 was examined using Autodock 4v2.6 software.
Estimation of the binding efficiencies indicated a comparable binding energy for
all four compounds (Table 1). However, calculated binding constants indicated a
higher affinity for the substances 4a and 4b (5.03
M and 10.97 M respectively) but lower for substances
4c and 4d (28.18 M and 47.44 M
respectively). A visual inspection of the complexes with Pin1 illustrated similar
conformations in three of the docked molecules but indicated interaction with
additional residues in the molecule containing the -tyrosine (substance
4d) head-group.
 Fig. 6.
Fig. 6.
The influence of the synthesised inhibitors on nuclear
localisation of p53. The structure of the four molecules were constructed using
the Alchemy program and saved in Brookhaven format (PDB). The crystal structures
of Pin1 (1PIN) was obtained from Brookhaven format (PDB). The location and
efficiency of binding of the molecules to Pin1 was estimated using the Autodock
4v2.6, the polar hydrogens were retained and partial charges added to the
proteins using the Gasteiger charges. The search space was limited to an area of
20 20 20 Å, centred around the hydroxyl group of Ser18
in the enzymatic site of Pin1.
Table 1.Approximation of binding of molecules to Pin1.
| Amino acid incorporated |
Binding energy |
Apoptosis |
| -Tyrosine |
–0.20 |
47.44 |
| -Tryptophan |
–0.22 |
10.97 |
| -Phenylalanine |
–0.22 |
28.18 |
| Naphthyl--alanine |
–0.23 |
5.03 |
| The structures of the four molecules were constructed using the Alchemy program
and saved in Brookhaven format (PDB). The crystal structures of Pin1 (1PIN) was
obtained from Brookhaven format (PDB). The location and efficiency of binding of
the molecules to Pin1 was estimated using the Autodock 4v2.6. For each enzyme, 25
ligand orientations (poses) were examined and ranked according to the
scoring-function and inhibition coefficient calculated. |
5. Discussion
Pin1 is involved in the regulation of a number of cellular processes,
particularly those associated with cancer. Pin1 overexpression is often
accompanied with the increased function of over 50 anti-apoptotic proteins and
repression of over 26 tumour suppressor proteins [34, 45, 46]. Pin1 has also been
reported to be a prominent mediator of epithelial-mesenchymal plasticity in
cancer cells [47]. As well as cancer cells, Pin1 has also been associated with
inflammatory responses during chronic disease such as atherosclerosis and
rheumatoid arthritis [48, 49]. The majority of the pro-inflammatory [50] and
cancer-related [51, 52] mechanisms stated above have also been firmly associated
with TF. The synchroneity between these proteins, and the recent demonstration of
the functional interaction of the proteins, suggests an inter-dependence which
may be exploited by using small molecules that can concurrently influence the
function of both of these other proteins. Such small molecules may include the
Pin1 inhibitors as discussed above, as well as the use of certain direct oral
anticoagulants (DOAC) which modify the TF-mediated signalling by inhibiting the
coagulation enzymes such as factor VIIa, and inducing the activation of
p53-mediated cell apoptosis [53, 54]. Moreover, to envision the usage of such
small molecules may confer distinct advantages to other strategies that are based
on the suppression of gene expression.
Recent studies have indicated differences in the cellular outcomes following
suppression of Pin1 expression, compared to strategies that inhibit of Pin1 [55].
Furthermore, variable outcomes have also been reported when using different
Pin1-inhibitors [55, 56]. Among the possible mechanisms for such distinct
influences, accessibility to different cellular compartments [57] or variability
in substrate recognition [58] may explain the underlying differences. The
function of Pin1 is closely regulated through a number of post-translational
modifications which indicates that Pin1 may be altered [34] in order to comply
with the required function when present in different cellular locations or
compartments. The ability of Pin1 to regulate the activity of TF and its release
from cells suggests a role in the dysregulation of procoagulant activity
associated with malignant compared to normal cells. Moreover, the close
association of TF with cancer progression and cellular survival indicates that
Pin1 may have an indirect homeostatic action as well as the commonly documented
functions.
The ability of Pin1 to interact with the phosphorylated form of cytoplasmic
domain of TF indicates the participation of membrane proximal/associated Pin1. In
this study we envisaged that it may be feasible to prepare a membrane-permeable
small-molecule inhibitor to specifically reduce TF activity and release in
microvesicles. Consequently, we prepared and evaluated four compounds based on
5-(p-methoxyphenyl)-2-methylfuran-3-carbonyl amide, with additional
aromatic head-groups to alter the binding capacity to Pin1. Compounds containing
-tryptophan (4b) and -tyrosine (4d) ‘head
groups’ were both capable of interfering with the binding of Pin1 to the
biotin-RKAGVGQSWKENpSPLNVS peptide. However, while compound 4d
suppressed the Pin1 activity towards the smaller Succ-ENpSPL-pNitroanilide
penta-peptide, compound 4b enhanced this activity. As an insight into
the possible differences, the in silico analysis of the interaction of
the compounds with Pin1 protein, indicated that despite the lower binding
affinity, compound 4d (containing -tyrosine) was capable of
interacting with Pin1 residues which were not engaged by any of the other
synthesised compounds (Fig. 6). However, the enhancement of Pin1 activity on
incubation with compound 4b, when tested against the pentapeptide but
not the TF-peptide, suggests that the inhibitors function by hindering the
interaction of Pin1 with the longer substrate.
The four compounds also exhibited different binding and functional potencies,
particularly towards TF activity and function. In agreement with the data
published by Kurakula et al., blocking of Pin1-TF interaction in cells,
using compounds 4b or 4d reduced the fXa generation activity on
the surface of cells. Moreover small, but significant decreases in cell surface
TF antigen, as well as the incorporation into microvesicles, were observed
following the incubation of cells with compound (4d) containing
-tyrosine (Fig. 3). However, in agreement with the enhancement of Pin1
isomerase observed with compound 4b (containing -tryptophan),
higher levels of TF incorporation and release were also observed following the
incubation of cells with compound 4b. Together, these data further
suggest that these compounds specifically interfere with the regulation of TF
function by Pin1. Therefore, while both compound 4b and 4d are
capable of preventing the approach of the longer peptides, the presence of acidic
tyrosine as the headgroup may also hinder the catalytic function of Pin1.
Isomerisation of TF on the cell surface by Pin1 is assumed to prolong its
presence on the cell surface [5] and allow its incorporation into microvesicles
[4]. Therefore, the inhibition of Pin1 would be expected to accelerate the
processing and endocytosis of TF into the cells [4, 20, 59]. However, it is
possible that compound 4b may also regulate other molecular components
involved in the formation as well as the translocation of proteins into
microvesicles. In contrast, our studies did not indicate any alterations in the
de novo expression of TF following supplementation of cells with either
of the compounds as has been previously reported [5]. One explanation for this
may arise from the inaccessibility of the nuclear compartment to the inhibitor
compounds and suggests that the synthesised compounds are limited to the
cytoplasmic/membrane regions [57]. Another possibility may be due to the reported
differences in the outcomes which are achieved following Pin1 knockdown or by
Pin1 inhibition [55]. Finally, the discrepancy may arise from differences in the
duration of incubation prior to mRNA quantification which was 6 h in our studies.
We previously demonstrated that disruptions in the ability of cells to moderate
and release TF can result in the activation of pro-apoptotic mechanisms, mediated
through src [25] and p38-MAPK [24] activation and leading to the nuclear
localisation of p53, upregulation of Bax and induction of cell apoptosis.
Consequently, we envisaged that the interruption of the regulation of TF
trafficking on the cell surface and reducing the rate of TF processing may
initiate these pro-apoptotic mechanisms. Therefore, in addition to the changes in
the cell-surface TF antigen and activity, the influence of the synthesised
compounds on the homeostatic mechanisms was investigated. In agreement with the
above hypothesis, incubation of cells with compound 4d (containing
-tyrosine) and to a lesser level, with compound 4b containing
-tryptophan resulted in reduced cell numbers, arising from cell
apoptosis as measured by DNA end-labelling and observable within 24 h. Incubation
of cells with compound 4a (containing 3-(2-naphthyl)--alanine)
also resulted in the reduction in cell numbers. However, none of the
pro-apoptotic mediators were altered and together with the non-specific release
of TF in microvesicles, these results indicate a non-specific cytotoxic effect by
compound 4a. Interestingly, these outcomes appeared to be specifically
mediated through TF since primary endothelial cells were not adversely affected.
Furthermore, compound 4d was able to promote nuclear localisation of p53
which may also arise from the retention of TF, although direct effects of Pin1 on
p53 localisation are well documented [30, 31, 32]. Finally, in agreement with the
above data, compounds 4b and 4d were able to induce the
expression of bax mRNA and Bax protein which explains the initiation of cell
apoptosis.
6. Conclusions
The regulation of the haemostatic and homeostatic functions of TF is a promising
direction and a potential therapeutic approach, particularly in cancer. In this
study, we aimed to assess the potential for synthetic small compounds, based on
previously known structures, to exert a regulatory effect on the procoagulant and
signalling properties of TF. Moreover, through targeting TF-mediated cellular
pathways, tumour cells but not endothelial cells were selectively eliminated. Of
the four synthesised compounds based on
5-(p-methoxyphenyl)-2-methylfuran-3-carbonyl amide, the inclusion of
-tyrosine as a head-group produced the most effective, functional and
applicable compound and constitutes a basis for further chemical formulations and
development for therapeutic exploration.
7. Author contributions
The study was designed by AM, ANB and CE, and the experimental work carried out
by OIA, MAM, SF and CE. The data were evaluated by OIA, ANB and CE and the
manuscript was prepared by OIA, ANB, AM and CE.
8. Ethics approval and consent to participate
Not applicable.
9. Acknowledgment
Not applicable.
10. Funding
The PhD studentship of OIA was co-sponsored the University of Abuja (Nigeria)
and the TETFund, Nigeria. No other external funding was received.
11. Conflict of interest
The authors declare no conflict of interest.
Abbreviations
Pin1, prolyl-protein cis/trans isomerase; TF, Tissue factor; fVII/X, factor
VII/X; fVIIa/Xa, activated factor VIIa/Xa; HDBEC, Human dermal blood
microvascular endothelial cells.
