
In Drosophila melanogaster, in response to developmental transcription factors, and by repeated initiation of DNA replication of four chorion genes, ovarian follicle cells, form an onion skin-type structure at the replication origins. The DNA replication machinery is conserved from yeast to humans. Subunits of the origin recognition complex (ORC) is comprised of Orc1, Orc2, and Cdc6 genes. While mutations of Orc1 and Orc2 and not Cdc6can be lethal, overexpression of these genes lead to female sterility. Ecdysone, is a steroidal prohormone of the major insect molting hormone 20-hydroxyecdysone that in Drosophila, triggers molting, metamorphosis, and oogenesis. To this end, we identified several ecdysone receptor (EcR) binding sites around gene amplification loci. We also found that H3K4 was trimethylated at chorion gene amplification origins, but not at the act1 locus. Female mutants overexpressing Lsd1 (a dimethyl histone H3K4 demethylase) or Lid (a trimethyl histone H3K4 demethylase), but not a Lid mutant, were sterile. The data suggest that ecdysone signaling determines which origin initiates DNA replication and contributes to the development. Screening strategies using Drosophila offer the opportunity for development of drugs that reduce gene amplification and alter histone modification associated with epigenetic effects.
In Drosophila melanogaster, different DNA replication systems are used during development (1- 5). During Drosophila oogenesis, endoreplication occurs in nurse cells, whereas both endoreplication and chorion gene amplification occur in follicle (fc) cells (1-5). The amplification of Drosophila chorion genes is necessary for eggshell formation, and mutations that disrupt amplification, such as those in cyclin E, orc2 (6, 7), chiffon (Dbf4) (8), humpty dumpty (9), and mcm6, cause female sterility. Orc2, orc5, and orc6 mutants have strong S-phase defects (7,10, 11). Surprisingly, dE2F1, dDP (12), dE2F2 (13, 14), Rbf (15), and the Myb complex (16,17) are necessary for this gene amplification process (18,19). We previously proposed that chorion gene amplification after repeated initiation of DNA replication at the origins occurs in response to developmental signals, initiated via transcription factors, in ovarian fc cells (20).
Orc1 is a large subunit of the origin recognition complex (ORC) and functions as the main subunit of the prereplication complex. In Drosophila, Orc1 levels are transcriptionally upregulated by E2F (21, 22) and downregulated by the anaphase promoting complex via proteolysis (23, 24). Neither Orc1 nor Orc2 in the salivary and ovarian fc cells is required for endoreplication (25). In the absence of Orc1, most amplification is diminished, whereas if Orc1 is overexpressed, DNA replication occurs throughout the nucleus. These results suggest that Orc1 is a limiting factor at least in some tissues (22). On the other hand, endoreplication in Drosophila does not require ORC for initiation, demonstrating that ORC-independent DNA replication can also occur (25).
In this study, we investigated where initiation of DNA replication begins and what triggers it at the chorion gene loci. in Drosophila. we report that regulation of signal transduction and DNA replication, especially with respect to the ecdysone receptor (EcR) and its cofactor TRR (26, 27), a histone H3 lysine 4 (H3K4) trimethylase, determines Orc1 loading at these loci.
Fly strains were maintained at 25°C on standard food. C323a-Gal4 driver and flies harboring UAS-EcR isoform transgene were obtained from the Bloomington Stock Center. Flies with orc1+-promoter-Orc1-GFP-9myc, UAS-Orc2-GFP-9myc, UAS-Orc1-GFP-9myc, UAS-Cdc6-GFP-9myc, or UAS-GFP were from M. Asano (Duke University Medical Center). UAS-Lsd1 was a gift from N. Dyson (HarvardMedical School, Boston, USA). UAS-Lid and UAS-Lid jmjC* were from R.N. Eisenman (Fred Hutchinson Cancer Research Center, Seattle, USA). Female sterility experiments were performed as described previously (28).
ChIP assays were performed mainly according to Kohzaki et al. (29, 30). Egg chambers from Orc1-GFP-9myc flies (23) were dissected from the ovaries of fattened flies in non-supplemented Grace’s medium (GIBCO-BRL). Formaldehyde was added to a final concentration of 2%, and cross-linking was allowed to proceed for 15 min at room temperature on a rotator. The cross-linking reaction was stopped by incubation with glycine at a final concentration of 0.125 mM for 5 min. The cross-linked egg chambers were washed twice with 1 ml of TBS, then twice with 1 ml of lysis buffer (30). The egg chambers were disrupted by sonication. Sonication and all postsonication procedures were performed as described previously (30). IgG (ab18413) and Myc antibody (9E10) was used in the ChIP assays. EcR and TRR antibodies were described previously (26, 27). H3K4me3 (ab8580) and H3K4me2 (ab7766) antibodies were purchased from Abcam (Cambridge, UK). Primers used were the same as those reported previously (29, 30).
Total RNA was isolated using Trizol Reagent (Invitrogen). Oligo dT primers and a Takara high fidelity RNA PCT kit (Takara, Kyoto, Japan) were used for generation of complementary DNA. Then, real-time PCR was performed using a SYBR Green I kit (Takara) and the Applied Biosystems 7500 real-time PCR system (Applied Biosystems, Foster City, CA, USA). RNA expression efficiencies decreased to 25% in every case (28).
Steroid hormones, including the prohormone ecdysone, play crucial roles during animal development. In Drosophila, ecdysone triggers molting, metamorphosis, and oogenesis through its effect on the gene expression network (28, 31-41). Ecdysone functions by binding to a nuclear receptor, EcR (42). EcR heterodimerizes with the retinoid X receptor ortholog Ultraspiracle (USP), which acts as a general heterodimer partner for the class of factors represented by EcR (39, 43-45). This heterodimer is required for binding to the ligands or their target DNA. The dimer activates EcR response gene expression by recruiting co-regulators. TRR, which is a histone methyltransferase capable of trimethylating H3K4, is required as a coactivator of EcR by modifying the chromatin structure at ecdysone-responsive promoters (26, 27). Ecdysone induces gene amplification at the Sciara coprophila DNA puff II/9A (46, 47).
Recently, we showed that Orc1 binds to ace1, ace, and ori-β directly (30) using flies with a single copy orc1 promoter orc1+-GFP-9myc transgene (Orc1-GFP9myc) (23). In eye imaginal disc of this transgenic fly, the behavior of Orc1-GFP9myc was essentially identical to that of ORC1. 1. Accumulation of Orc1-GFP9myc prior to CycB in late G1 or early S phase, 2. Persistence of both Orc1-GFP9myc and CycB throughout G2, 3. Removal of Orc1-GFP9myc from chromatin during M phase upon accumulation of PH3, 4. Disappearance of all three antigens (Orc1-GFP9myc, CycB and PH3) upon entry into the subsequent G1 (24) but not in ovary, especially in follicle cells. We previously showed that the transgene expressed in ovary using ChIP assay (30). Here we considered this issue using microscopes histologically.
In this Orc1-GFP9myc fly, Orc1-GFP was expressed similarly to endogenous Orc1 (Figure 1A and C) and localized to fc cells (Figure 1B and D). We asked whether response elements are found around the gene amplification loci. We detected several EcR putative binding sites (Figure 2A). In these fly ovaries, we performed a ChIP assay using EcR-C monoclonal antibody and TRR polyclonal antibody (26, 27). We detected signals at ace3, ori-β, and ace1 (Figure 2B and C). The amounts of ace1, ace3, and ori-β PCR products obtained using EcR, TRR, H3K4me3, Myc (for Orc1) and IgG antibodies relative to those obtained using H3K4me2 antibody were considerably different (t-test, p < 0.05) (48). Also, the amounts of ace1, ace3, and ori-β PCR products obtained using EcR, TRR, Tri-Me, and Myc (for Orc1) antibodies were not statistically different (t-test, p > 0.05) (48) (Figure 2). These data suggest that EcR, TRR, and Orc1 might form a complex for initiation of DNA replication.
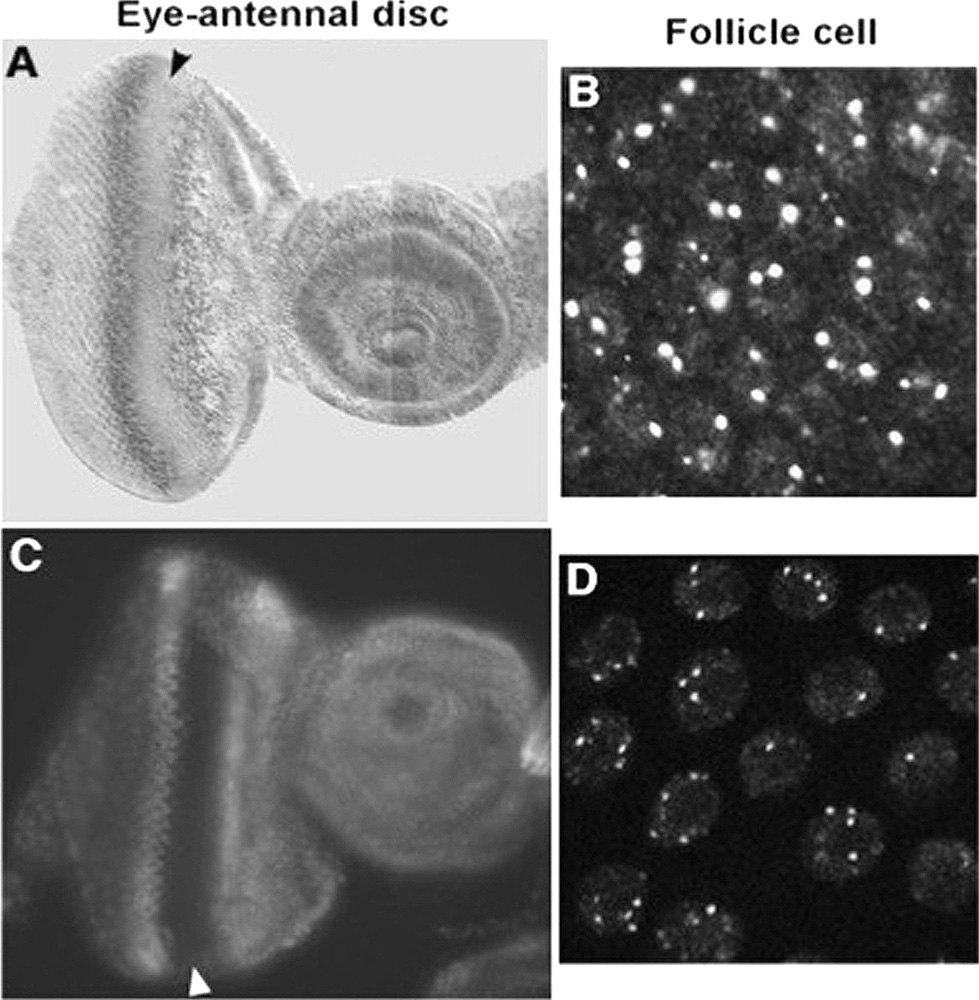 Figure 1
Figure 1Distribution of Orc1 and Orc1-3HA-GFP. (A, B) Antibody straining reveals the distribution of endogenous Orc1 using anti-Orc1. (C, D) Fluorescent signals from Orc1-GFP9myc driven by the orc1 promoter. This transgene was in a wild-type background. In A and C, arrowheads show the morphogenetic furrow (MF) in eye-antennal imaginal discs. MF migrates from the posterior (the left side) to the anterior (the right side). Most eye disc cells first undergo one synchronous cell cycle and then enter a prolonged G1/G0 phase. In B and D, dots indicate gene amplification loci associated with Orc1 and Orc1-3HA-GFP.
 Figure 2
Figure 2(A) Schematic representation of EcR-binding sites. Asterisks indicate putative binding sites. The USP-binding site was reported previously (60-61). (B) Association of EcR, TRR, H3K4me3, H3K4me2, and Orc1-GFPmyc with chorion gene elements in vivo. ChIP assays were performed as described in Methods with anti-EcR, anti-TRR, anti-H3K4me3, anti-H3K4me2, and anti-myc antibody. DNA was amplified using PCR primers specific to ace1, ace3, ori-β, and act5C 5’UTR as described previously (29) in Figure 2A. These primers were also used to amplify DNA isolated from whole cell extracts before immunoprecipitation (WCE). The experiments were repeated to confirm reproducibility. The same PCR products were loaded onto separate gels for each primer set. The samples derived from the same experiment and the gels were processed in parallel. “– “ was shown that IgG was used as negative control. (C) Quantitation of ChIP assays was performed at the times as described (N) using NIH image (Image J). The amounts of PCR products obtained from 10% input of WCE were taken to be equal to 1.0. As negative control, IgG was used. As a sample, about ori-β PCR products, the product of EcR, TRR, Tri-Me and Orc1 was compared with that of Di -Me. *; T test, p<0.05. The amounts of ace1, ace3, and ori-β PCR products obtained using EcR, TRR, Tri-Me, and Myc (for Orc1) antibodies were not statistically different (t-test, p > 0.05).
Four EcR isoforms have been isolated (41, 49), and each has a tissue-specific function during development. We overexpressed each EcR in fc cells. Overexpression of all EcRs except EcR.B1 led to female sterility, but overexpression of the EcR mutant F645A, which did not have transcription activity, did not (Figure 3A). Therefore, ecdysone may regulate gene amplification directly through transactivation.
 Figure 3
Figure 3(A) Overexpression of dominant negative forms of EcR led to female sterility. Females with the c323a-Gal4 driver were crossed with male flies harboring a UAS-EcR isoform transgene (a responder). The progeny (males or females) having the indicated responder and c323awere crossed with wild-type Canton-S (CS) flies. All responders were tested: if no bar is visible, there were no progeny with wild-type growth rates and rates of larvae emergence. The experiments were performed several times (N). The numbers of progeny from CS (male) × c323a expressing EcR families (female) are 100% (blue bars in Figure 3A). (B) Overexpression of Lsd1 and Lid, but not Lid jmjC*, led to female sterility. Assays were performed as described in Figure 3A with flies harboring UAS-Lsd1, UAS-Lid, or UAS-Lid jmjC* transgenes. The experiments were performed several times (N). The numbers of progeny from CS (male) × c323a expressing Lsd, Lid, or Lid jmjC* families (female) are 100% (blue bars in Figure 3A).
We next asked whether H3K4 is converted to the trimethylated form (H3K4me3) by TRR. TRR was identified as a Set domain protein in Drosophila and is highly homologous to Drosophila TRITHORAX protein and to human ALL-1/HRX. TRR mutants trr1 and trr3 are embryonic lethal 8 (26). H3K4me3 is associated with transcriptionally active genes in eukaryotes (50, 51). We checked the localization of H3K4me3 around the gene amplification loci. Actin 5C is transcribed during early embryogenesis, and its 5’UTR is trimethylated, not dimethylated. We found H3K4me3 around ace3, ori-β, and ace1. H3K4 was also dimethylated (Figure 2B and 2C). These data showed that the gene amplification locus encoding genes for choriogenesis is euchromatic. Gene amplification was be induced by ecdysone signaling (Figure 2B and C). We observed a correlation between gene amplification-associated H3K4me3 and TRR loading in fc cells (Figure 2B and 2C). To investigate the biological significance of H3K4me3, we overexpressed H3K4 demethylase Lsd1 or Lid in fc cells. Many groups have isolated H3K4 demethylases and trimethylases in the same species (52-60). In Drosophila, Lsd1 is a dimethyl histone H3K4 demethylase (58) and Lid is a trimethyl histone H3K4 demethylase (61-63). Lsd1 or Lid overexpression in females caused sterility (Figure 3A and 3B). By contrast, females expressing Lid jmjC*, which has a mutation in its active site, or Cdc6 or GFP (28) were normal. These data suggest that H3K4me3 is essential for gene amplification.
Orc1 is the key player in initiation because it binds to chromatin via its BAH domain and is degraded in a cell cycle-dependent manner (21, 30, 35, 64). For functional ORC formation, Orc1 requires the Orc core complex, Orc2–5 (9, 29, 65, 66). Orc1 is loaded on gene amplification origins (Figure 2A and B). These data suggest that developmental signals can regulate gene amplification. We speculate that gene amplification this reaction is coupled to transcription.
In summary, the data suggest that ecdysone signaling determines which origin initiates DNA replication (Figure 4).
 Figure 4
Figure 4Schematic representation of the putative mechanism by which ecdysone signals could regulate chorion gene amplification. EcR: Ecdyson receptor, USP: RXR ortholog, Ultraspiracle, TRR: Histone H3K4 methyltransferase, ORC: Origin recognition complex
Here, we showed that EcR regulates chorion gene amplification through the activity of the H3K4 trimethylase TRR. Overexpression of H3K4 demethylases Lsd1 and Lid, but not the jmjC mutant Lid jmjC* (63), led to female sterility. Because EcR, TRR (26, 30), and Orc2 mutants experience a growth defect before chorion gene amplification, EcR signaling may direct gene amplification. The heterodimer partner USP was identified originally as chorion factor 1, which binds to the chorion s15 cis-regulatory element (2, 67, 68). This elementincludes ori-β and the putative EcR-binding sites (Figure 2A). We propose that EcR-USP-TRR binds to the region between S18 and ori-β.
We previously showed that transcription factors regulate ORC loading and initiation of DNA replication via chromatin modifications in S. cerevisiae (69) and Drosophila (30). The initiation of gene amplification is linked to histone H3 and H4 hyperacetylation and H1 phosphorylation in Drosophila (70). Indeed, in Drosophila fc cells, binding of Rpd3 or Polycomb proteins to origins decreases their initiation activity, whereas binding of the Hat1 homolog, Chameau acetyltransferase, increases origin activity (1). The assay used an artificial technique because of fusing these proteins to the Gal4 DNA binding domain. Then, the situation in vivo remains unclear. dE2F-dDP-Rbf interacts with DmOrc and dE2F1, and DmOrc binds to chorion gene amplification loci in vivo (15). Because the authors did not identify the E2F-binding site or the mutation disrupting the interaction, it is unknown whether they bind directly or indirectly.
EcR isoforms are functionally distinct. When early genes are expressed in tissues, the EcR-A isoform is dominant. The other EcR isoform, EcR-B1, is the predominant isoform in both the imaginal and larval cells of the larval midgut (71). What induces differences in expression and function? Bender et al. suggested that tissue-specific coactivators, such as TRR, may provide the link between the transcription machinery for a given gene and a particular EcR isoform (31). If this is the case, it would be the coactivator that determines which EcR isoform is used to activate the gene. These might be akin to the plethora of putative coactivators recently found for vertebrate nuclear receptors (31, 72) (Figure 4).
In mammals, many transcription factors are proto-oncogenes, including c-Jun, c-Myb, and c-Myc (20, 73). Their oncogenicity is thought to be due to dysregulation of the transcription that they promote. By contrast, we speculate that it is the dysregulation of replication caused by the multifunctionality of these transcription factors that contributes to their oncogenic potential. This speculation is supported by previous reports (20, 74). c-Jun homologhas been shown to regulate ORC loading in S. cerevisiae (69) and a c-Jun ortholog, Gcn4, promotes ORC loading. In Drosophila, a myb gene mutant induces a defect in S-phase progression in several tissues. c-Myc modulates DNA replication origin activity through the regulation of Cdc45 loading (74, 75). We previously propose that the DNA replication machinery contributes to development (28, 77). Changes in the space- and time-controlled process of development can lead to dys-regulated DNA synthesis, checkpoint activation, genomic instability, and/or cell death.
In America, 12.1% of women aged 15–44 have impaired fecundity and 7.3 million (12.0%) have never used infertility services, and 6.7% of married women aged 15–44 are infertile (77).
The life cycle of Drosophila is short. Screening strategies using these flies could potentially lead to the development of drugs for the treatment of sterility.
We dedicate this paper to M. Asano. We thank N. Dyson (Harvard Medical School, Boston, USA), R.N. Eisenman (Fred Hutchinson Cancer Research Center, Seattle, USA), and M. Asano for flies harboring UAS-Lsd, UAS-Lid, and UAS-Lid jmjC*. We thank M. Asano for technical advice and gifts of orc1+-promoter-Orc1-GFP-9myc, UAS-Orc2-GFP-9myc, UAS-Orc1-GFP-9myc, UAS-Cdc6-GFP-9myc, and UAS-GFP. We dedicate this paper to M. Asano (The Ohio State University). We thank Dr. Tadashi Uemura (Kyoto University) and members of his laboratory for their dedicated support and helpful assistance. This work was partially supported by the Japanese Leukemia Research Fund. H.K. was supported by a KIT VL grant and the Memorial Fund on the 44th Annual Meeting of the Japan Society for Clinical Laboratory Automation and The Motoo Kimura Trust Foundation for the Promotion of Evolutionary Biology. This manuscript has been released as a Pre-Print (78).