
PolyGalacturonase Inhibiting Proteins (PGIPs) are leucine rich repeat pathogenesis-related (PR) cell wall proteins, which interact and inhibit the PolyGalacturonase (PG), an enzyme secreted by the pathogen to degrade pectin. Interaction of PGIP with PG limits the vulnerability of PG by the activation of host defense response against pathogenic attack. Erwinia is gram-negative soft rot bacteria responsible for rhizome rot disease in banana and many other crop plants. The interaction of PG with PGIP is one of the crucial steps for plant-pathogen interaction. To study the molecular mechanism of PR proteins, we employed molecular modelling, protein-protein docking and molecular dynamics simulations of banana PGIP (bPGIP) with Erwinia carotovora PG (ecPG). Further, insilico site-directed mutagenesis was performed in Phaseolus vulgaris PGIP (pvPGIP2) to elucidate the interaction with ecPG. Docking and simulation studies divulge that binding of bPGIP and PvPGIP2 with active site residues of EcPG induces structural changes and thereby inhibit the enzyme. This study provides a unique insight into PG-PGIP interaction, which may help in the development of bacterial soft-rot resistant banana cultivars.
Crop plants in agro-ecosystem are continuously facing the danger of pathogenic infections from fungi, bacteria, viruses, and nematodes (1). However, the degree of plant defense against pathogens depends on its recognition ability of pathogen and host-plant defense response cascade (2). The plant system is having cell surface receptors which are sensitive to pathogen associated molecular patterns (PAMPs) and activates cascade of defense related activities including innate immune responses against invading pathogens (3, 4). PAMPs are microbial molecules like glycans and glycol-conjugates which stimulate innate immunity in the host to protect from infection by recognizing few conserved non self-molecules. PolyGalacturonase Inhibiting Protein (PGIP) is a type of pathogenesis-related (PR) plant protein present at the cell surface and intracellular spaces of the plant cells. It interacts and inhibits the enzyme PolyGalacturonase (PG) which is secreted by the pathogen such as Agrobacterium vitis, Pectobacterium carotovora, Xylella fastidiosa and Botrytis cinerea, to degrade heteropolysaccharide pectin (5, 6). The PG enzymes secreted by pathogen Erwinia spp. are known to infect more than 100 plant species in several families. It is a lethal pathogen in the field as well as in storage conditions and are responsible for heavy economic losses. Various in vitro studies showed that the interaction of plant PGIP with PG of pathogen limits the vulnerability of PG and results in the accretion of elicitor-active oligogalacturonides to activate a quick defense response against pathogenic attack (7-9).
PGIPs are the important member of leucine-rich repeat (LRR) proteins super-family (10). These LRR proteins show a pivotal role in plant resistance with several resistance genes (11). The LRR proteins are structural motif having very specific roles in the protein-protein interaction (PPI) of host-pathogen which played immunity functions in plants. PGIPs from the various plants differ in inhibitory action against PG. PGIPs from the same plant species inhibit PGs from different bacterial species or multiple host infecting bacteria with variable aggressiveness or virulence potential. PolyGalacturonases that degrades homogalacturonan (HGA) polymer, a key constituent of pectin in plant cell walls, by hydrolysis of the glycosidic bonds during early stages of infection (12). The 1, 4 linked alpha-D-galactosyluronic acid is a linear homopolymer of HGA present in the plant cell, and is the first line of defense to the invading pathogen (13-16). Protein-protein interaction (PPI) between PG–PGIP complexes are a typical system in the background of plant–pathogen interaction mechanism (14). The experimental three-dimensional structures of various PGs are available in RCSB Protein Data Bank (PDB) to date (17-19), but, in case of PGIP only crystal structure of Phaseolus vulgaris (PvPGIP) is available till date in PDB (20).
Molecular approach can provide clue for better genetic control of plant resistance reducing damage caused by pathogens (21). Significant biotechnological achievements have been reported in the field experiments with several isoforms of PGIP from diverse group of plants. The resistance in plants was successfully achieved by over expression of PGIP from various plants against fungal pathogens (9, 22-25). A critical mining of literature indicates that very few studies have been performed against PGs of insects and bacteria till date (26) and interestingly, there is no report of inhibition or resistance achieved against bacterial prominent soft rot pathogen like Erwinia spp. PGIP protein has been found very promising in increase in breeding efficiency of banana against various biotic stresses have been reported by radiation mutagenesis (27, 28), chemical mutagenesis (29, 30) and genome editing (31).
Prevalent chemical method of disease control, for example, use of foliar spray of copper oxychloride and streptomycin sulphate has several environmental and health issues (32). Use of antibiotics like streptomycin has very high risk/potential threat of antibiotic resistance development (33) which is also noticed in bacterial phytopathogens (34). Though traditional method of banana germplasm improvement has been successful but still it has limitations in resistant cultivar development (35). All these issues necessitate banana cultivar development using alternative strategies at molecular level.
In host-pathogen recognition of banana and gram-negative bacterial pathogen Erwinia PG-PGIP interaction plays a pivotal role. Modelling of 3D structure of PGIP of banana and molecular dynamics simulation studies of PG-PGIP complexes can provide deep insights into the molecular mechanism of recognition. In such study, in silico mutation of PGIP can be a prudent, rapid and cost effective approach having advantage to predict most effective artificially created allele fetching durable resistance in plants.
The present work aims at host pathogen interaction studies having 3D model of banana PGIP and molecular dynamics simulation studies to reveal critical amino acids involved in molecular recognition with PG of bacterial pathogen, Erwinia spp. The study further evaluates in silico site-directed mutagenesis of selected critical amino acids of PGIP peptide as model approach to predict where desirable changes can be made to inhibit the host pathogen recognition, which is required in development of soft rot resistant banana genotype.
The 3D structures of bPGIP proteins are not available in PDB. Thus, the amino acid sequence of the PGIP (Musa AAB Group) was fetched from NCBI protein database (ADQ38901.1). BLASTp search was executed against the PDB to find appropriate template protein structures for homology modelling (36). The best template (PDB ID: 1OGQ) showing maximum identity and high score as well as lower e-value was selected for building 3D structures. The sequence alignment between target and template was performed using MultAlin program. MODELLER9v19 was used for 3D structure prediction (37, 38). Out of 20 models (3D structures) resulted from MODELLER, one 3D structure was selected and subjected to further stereo-chemical properties check to get the deviations from the standard bond length, dihedrals and non-bonded atom-atom distances. The 3D structure models of PGIP were further calculated based on discrete optimized protein energy (DOPE) scores. The model with lowermost DOPE scores was selected for further refinement using GROMACSv5.1 simulation package. We performed in silico site-directed mutagenesis in PvPGIP2 by using the mutate_model command of MODELLER in which Q224 and V152 were replaced with the corresponding residues Lys and Gly, respectively.
The stereo-chemical quality of generated 3D structure was verified using SAVES server, ProSA and ProQ. MolProbity tool was used to calculate bond length and bond angle of the 3D model. The stereo-chemical properties and Ramachandran plots was analysed using Procheck analysis. Furthermore, the Z-score, packing defects, bump score; radius of gyration and deviation of Y angles of the selected model was observed using VADAR, GeNMR and PROSESS web-servers. Additionally, to evaluate the accuracy of the model, the structural superimposition of corresponding Cα trace of the predicted structure over template structure (PDB ID: 1OGQ) was executed using PyMOL (The PyMOL Molecular Graphics System, Version 2.0 Schrödinger, LLC.).
The 3D structure of PG of Erwinia carotovora was retrieved from PDB (PDB ID: 1BHE) and modelled 3D structure of Musa PGIP and mutated PvPGIP2 were used for protein-protein interaction (PPI) studies. The active residues in both the proteins PGIP and PG for PPI studies were identified and used for docking via HADDOCK (High Ambiguity Driven biomolecular DOCKing) server and Patchdock. HADDOCK is an information-driven docking approach which uses non-structural experimental information to perform docking through a rigid-body minimization of energy, semi-flexible in addition to water refinement stages. PatchDock, a geometry-based molecular docking algorithm was used for enzyme (ecPG)-inhibitor (bPGIP and mutated PvPGIP2) docking with a clustering RMSD of 4Å. The complexes of bPGIP-ecPG and mPvPGIP2-EcPG docked candidates obtained from Patchdock were passed to FireDock that refined and scored them according to an energy function. Twenty solutions, out of about 35 predicted PG-PGIP complexes, were sorted conferring to their geometric shape. The complementarities scores were analysed for the identification of involved residues in the protein-protein interface. The complexes of the best cluster were further analysed by BIOVIA Discovery Studio Visualizer (Dassault Systèmes BIOVIA, BIOVIA DSV, 4.5, San Diego: Dassault Systèmes, 2017) to choose the best complex.
To study the dynamical properties, MD simulations were performed for modelled PGIP, PG (1BHE), PGIP-PG complexes (bPGIP-1BHE, PvPGIP2-1BHE) and mutated complex (mutated PvPGIP2-1BHE) through GROMACSV5.1 package. For topology building, CHARMm force fields were used in cubic boxes with TIP3P water model; periodic boundary conditions were applied by keeping the distance between the edge of the box and the protein as 1nm. The protonation states of all the ionisable amino acids were determined at pH 7.0. The structures were solvated and sodium counter ions (0.15 M NaCl) were added to neutralize the overall charge of the systems. Energy minimization of the solvated systems were performed using conjugate gradient and steepest descent algorithm until maximum force reached less than 1000 kJmol-1nm-1. After energy minimisation, each system was subjected to NVT and NPT ensemble for equilibration at 1000 ps. LINear Constraint Solver (LINCS) algorithm was implied to constrain all bond lengths. Electrostatics was calculated by using Particle mesh Ewald method with a cut-off distance of 0.9 nm. Finally the equilibrated systems were subjected to final MD run of 50 ns at 300 K.
The intrinsic stability parameters i.e., backbone root mean square deviation (RMSD), radius of gyration, solvent accessible surface area (SASA), inter-molecular Hydrogen-bond and from each trajectory was analysed using utility toolkits of GROMACS and 2D graphs were plotted using Grace (http://plasma-gate.weizmann.ac.il/Grace/). The interaction images were plotted using BIVIA DSV and PyMOL.
Essential Dynamics or Principal Component Analysis (PCA) was executed to explore and understand the global motion of the atomic coordinates of PGIP and 1BHE as well as of mutated PvPGIP2 and 1BHE during MD simulations. PCA systemically decreases the dimensionality of a complex and also can describe the collective and overall motion of the protein system. The first principal component (PC1) includes the largest Root-Mean-Square Fluctuation (RMSF). The fluctuations in the system are due to the correlation between the motion of the particles, where the motion of the particles directly proportional to the function of the protein. The sum of fluctuations of the collective motions per atom is given by the eigenvalues thus, these gives the total motility associated with the eigenvector. PCA calculations were performed by gmx covar and gmx aneig utility functions of GROMACS. Our investigation was limited to Cα atoms, as it is less perturbed by statistical noise and provides a substantial characterization of the essential space motion.
The structure of bPGIP protein (comprised of 286 amino acids) was predicted by homology modeling approach. BLASTp search against PDB with the maximum identity (identity=49% and similarity =67%), high score and lower e-value resulted 1OGQ as the best template for the homology modeling. The pair-wise sequence–structure alignment has been displayed in Figure 1. The alignment was considered by few insertions and deletions in the loop regions. Modelling was carried out from 23 to 273 residues after that a refinement of the model was done through energy minimization by means of CHARMm36 force field using GROMACS (39). The final optimized structure of PGIP of Musa protein has been displayed in Figure 2A.
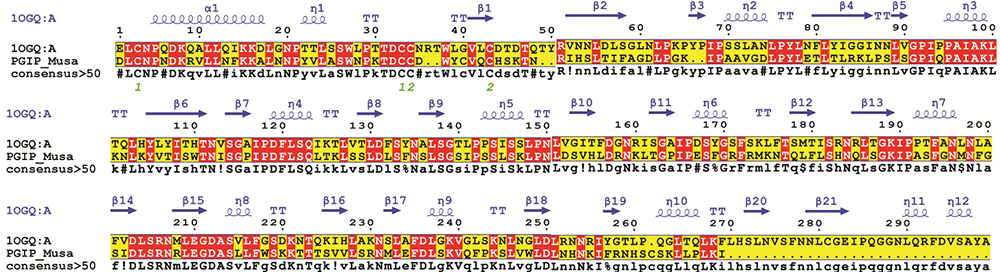 Figure 1
Figure 1
Pair-wise sequence structure PGIP of banana and its closest structural homolog i.e., crystal structure of PvPGIP (PDB ID: 1OGQ_A) was constructed using MultAlin. The image was rendered using ESPript. The secondary structural elements were obtained from the crystal structure 1OGQ. The α-helices, η-helices, β-sheets and strict β-turns are denoted α, η, β and TT respectively. Similar amino acids are highlighted in boxes, and completely conserved residues are indicated by white lettering on a red background.
 Figure 2
Figure 2
Three-dimensional predicted structure of the protein. (A) Three-dimensional architecture and structural imposed view of the modeled bPGIP (displayed in green) with the crystal structure of PvPGIP (1OGQ_A shown in Red). (B) The phi and psi distribution of amino acids in the available zones of Ramachandran plot obtained using PROCHECK. (C) ProSA-Web Z-score profile the modeled bPGIP a z-score of −7.4, comparable to those of experimentally determined structures of similar size.
The electrostatic surface potential of the modelled bPGIP was calculated using APBS plugin of PyMOL as shown in Figure 3. Looking at the concave surface of the bPGIP, it can be ascertained that negatively charged residues dominates over positively charged amino acids.
 Figure 3
Figure 3
Electrostatic surface potential of bPGIP. (A) The side view of bPGIP. (B) The rotated view displaying the charge distribution of amino acids of the concave binding surface of bPGIP.
The stereo-chemical properties of the modelled structure of PGIP of Musa was analysed using PROCHECK. It was observed that the phi/psi angles of most of the residues (91.2%) fell in the most favored regions (Figure 1B). The inclusive PROCHECK G-factor for the predicted structure was -0.26. The model was furthermore validated using ERRAT graph. The overall quality factor (74.11) recommends that the quality of model was good (a score of >50 for a rational model). PROSA Z-score value was found to be negative (−7.46) for the predicted model (Figure 1C), which was quite closer to that of the template (−8.2), indicating the consistency of the model. A quality of model was also established from VERIFY 3D server as 95.24% of residues presented a score > 0.2. Results of WHATCHECK also indicated the correctness of the model. On the basis of these diverse validation experiments, it was determined that the modelled structure has rationally good quality. The bond length and bond angles analysis carried out by MolProbity which evaluates the model quality both at global and local levels for proteins. By PROSESS, mean Chi-1 standard deviation was found to be 13.45 and 12.59 which was also found to be in the defined range. ProQ is a neural network based predictor that based on a number of structural features predicts the quality of a protein model. Two quality measures are predicated LGscore and MaxSub. The proQ results for bPGIP model was 6.45 (LGscore) and 0.478 (MaxSub) which confirmed the good quality of the model as LGscore above 4 is considered as extremely good model. The structural superimposition of Cα trace of the predicted structure over template structure 1OGQ (Figure 1A, lower panel) resulted in a root mean square deviation (RMSD) of 0.34 Å using PyMOL specifies a valid structure of the model.
The final refined model of PGIP and 3D structure of Erwinia PG (1BHE) were used as input in LIGSITE to find the active residues for both the proteins. In bPGIP, active residues were Leu24, Cys33, Cys34, Tyr37, Cys38, Cys41, Arg47, Ile48, Thr52, Ile53, Phe54, Gly56, Ile87, Ile91, Val100, Ile102 and Leu115 whereas, in case of 1BHE active residues were identified as Ser3, Lys210, Asn211, Asn239, Asn277, Arg280, Lys306, Val309, Asp311 and Tyr314. These residues were used as input in HADDOCK server for studying PPI. HADDOCK clustered 129 structures in 17 clusters representing 64.5% of the water-refined models. Clustered complex with a Z-score -2.7 was further used as input in DSV for further interaction analysis. A similar procedure was followed for PvPGIP2-1BHE and mutated PvPGIP2 and 1BHE complex. The active residues of PvPGIP2 and mutated mPvPGIP2 along with 1BHE active residues were submitted in HADDOCK server. HADDOCK clustered 41 structures in 8 clusters which represent 20.5 % of the water-refined models. Clusters with a Z-score of -1.4 were selected and used for further analysis by DSV. Inter-molecular analysis revealed a total of 14 H-bonds in PvPGIP2-1BHE complex and 34 bonds (19 hydrogen bonds, 6 electrostatic bonds, and 9 hydrophobic bonds) in mutated pvPGIP2-1BHE complex. Details of these intermolecular-interactions have been listed in Tables 1-3. The interaction analysis of these (PvPGIP2-1BHE and mutated PvPGIP2-1BHE) complexes provides a strong clue for production/cultivation of genotype having resistant against rhizome and soft rot caused by Erwinia spp. Furthermore, the interaction study of bPGIP-EcPG provides an effective example of targeting PGs of bacterial soft rot pathogens by PGIP.
| S. No | Name | Distance | Category | Type | From | From Chemistry | To | To Chemistry |
|---|---|---|---|---|---|---|---|---|
| 1 | B:LYS338:HZ1 - A:GLU73:OE1 | 1.65134 | Hydrogen Bond;Electrostatic | Salt Bridge;Attractive Charge | B:LYS338:HZ1 | H-Donor;Positive | A:GLU73:OE1 | H-Acceptor;Negative |
| 2 | B:LYS370:HZ3 - A:ASP35:OD1 | 1.57898 | Hydrogen Bond;Electrostatic | Salt Bridge;Attractive Charge | B:LYS370:HZ3 | H-Donor;Positive | A:ASP35:OD1 | H-Acceptor;Negative |
| 3 | A:LYS98:NZ - B:ASP364:OD1 | 5.07459 | Electrostatic | Attractive Charge | A:LYS98:NZ | Positive | B:ASP364:OD1 | Negative |
| 4 | B:LYS142:NZ - A:ASP57:OD1 | 4.68447 | Electrostatic | Attractive Charge | B:LYS142:NZ | Positive | A:ASP57:OD1 | Negative |
| 5 | A:TYR22:HH - B:VAL143:O | 2.26973 | Hydrogen Bond | Conventional Hydrogen Bond | A:TYR22:HH | H-Donor | B:VAL143:O | H-Acceptor |
| 6 | A:TYR37:HH - B:ASN277:OD1 | 2.83369 | Hydrogen Bond | Conventional Hydrogen Bond | A:TYR37:HH | H-Donor | B:ASN277:OD1 | H-Acceptor |
| 7 | A:LYS79:HZ2 - B:GLY274:O | 1.87316 | Hydrogen Bond | Conventional Hydrogen Bond | A:LYS79:HZ2 | H-Donor | B:GLY274:O | H-Acceptor |
| 8 | A:LYS79:HZ3 - B:THR276:OG1 | 1.72395 | Hydrogen Bond | Conventional Hydrogen Bond | A:LYS79:HZ3 | H-Donor | B:THR276:OG1 | H-Acceptor |
| 9 | B:LYS142:HZ3 - A:ASP57:O | 1.63442 | Hydrogen Bond | Conventional Hydrogen Bond | B:LYS142:HZ3 | H-Donor | A:ASP57:O | H-Acceptor |
| 10 | B:LYS306:HZ2 - A:CYS38:O | 2.26925 | Hydrogen Bond | Conventional Hydrogen Bond | B:LYS306:HZ2 | H-Donor | A:CYS38:O | H-Acceptor |
| 11 | B:TYR314:HH - A:VAL23:O | 2.31979 | Hydrogen Bond | Conventional Hydrogen Bond | B:TYR314:HH | H-Donor | A:VAL23:O | H-Acceptor |
| 12 | B:LYS316:HZ3 - A:ALA25:O | 1.83285 | Hydrogen Bond | Conventional Hydrogen Bond | B:LYS316:HZ3 | H-Donor | A:ALA25:O | H-Acceptor |
| 13 | B:GLN368:HE21 - A:CYS34:O | 2.06204 | Hydrogen Bond | Conventional Hydrogen Bond | B:GLN368:HE21 | H-Donor | A:CYS34:O | H-Acceptor |
| 14 | B:GLN368:HE22 - A:TRP36:O | 2.1108 | Hydrogen Bond | Conventional Hydrogen Bond | B:GLN368:HE22 | H-Donor | A:TRP36:O | H-Acceptor |
| 15 | B:ALA139:CA - A:ASP57:OD1 | 3.4354 | Hydrogen Bond | Carbon Hydrogen Bond | B:ALA139:CA | H-Donor | A:ASP57:OD1 | H-Acceptor |
| 16 | B:LYS338:CE - A:THR74:OG1 | 3.31255 | Hydrogen Bond | Carbon Hydrogen Bond | B:LYS338:CE | H-Donor | A:THR74:OG1 | H-Acceptor |
| 17 | B:THR276:CG2 - A:PHE54 | 3.51849 | Hydrophobic | Pi-Sigma | B:THR276:CG2 | C-H | A:PHE54 | Pi-Orbitals |
| 18 | A:TYR37 - B:TYR314 | 5.16823 | Hydrophobic | Pi-Pi T-shaped | A:TYR37 | Pi-Orbitals | B:TYR314 | Pi-Orbitals |
| 19 | A:ALA25 - B:LYS316 | 3.97328 | Hydrophobic | Alkyl | A:ALA25 | Alkyl | B:LYS316 | Alkyl |
| 20 | A:TYR22 - B:LYS145 | 4.57981 | Hydrophobic | Pi-Alkyl | A:TYR22 | Pi-Orbitals | B:LYS145 | Alkyl |
| 21 | A:TYR37 - B:VAL342 | 5.33111 | Hydrophobic | Pi-Alkyl | A:TYR37 | Pi-Orbitals | B:VAL342 | Alkyl |
| 22 | B:HIS251 - A:ALA55 | 4.65041 | Hydrophobic | Pi-Alkyl | B:HIS251 | Pi-Orbitals | A:ALA55 | Alkyl |
| S. No | Name | Distance | Category | Type | From | From Chemistry | To | To Chemistry |
|---|---|---|---|---|---|---|---|---|
| 1 | A:LYS225:HZ1 - B:ASP311:OD2 | 1.62127 | Hydrogen Bond ;Electrostatic | Salt Bridge; Attractive Charge | A:LYS225:HZ1 | H-Donor; Positive | B:ASP311:OD2 | H-Acceptor; Negative |
| 2 | A:LYS268:HZ1 - B:ASP364:OD1 | 1.6792 | Hydrogen Bond ;Electrostatic | Salt Bridge; Attractive Charge | A:LYS268:HZ1 | H-Donor; Positive | B:ASP364:OD1 | H-Acceptor; Negative |
| 3 | B:LYS142:HZ2 - A:ASP294:OD1 | 3.1038 | Hydrogen Bond ;Electrostatic | Salt Bridge; Attractive Charge | B:LYS142:HZ2 | H-Donor; Positive | A:ASP294:OD1 | H-Acceptor; Negative |
| 4 | B:LYS285:HZ1 - A:ASP131:OD1 | 1.64033 | Hydrogen Bond ;Electrostatic | Salt Bridge; Attractive Charge | B:LYS285:HZ1 | H-Donor; Positive | A:ASP131:OD1 | H-Acceptor; Negative |
| 5 | B:LYS316:HZ3 - A:ASP203:OD2 | 1.56125 | Hydrogen Bond ;Electrostatic | Salt Bridge; Attractive Charge | B:LYS316:HZ3 | H-Donor; Positive | A:ASP203:OD2 | H-Acceptor; Negative |
| 6 | B:LYS349:HZ2 - A:ASP56:OD1 | 1.61466 | Hydrogen Bond ;Electrostatic | Salt Bridge; Attractive Charge | B:LYS349:HZ2 | H-Donor; Positive | A:ASP56:OD1 | H-Acceptor; Negative |
| 7 | A:ARG183:NH1 - B:GLU318:OE1 | 4.22015 | Electrostatic | Attractive Charge | A:ARG183:NH1 | Positive | B:GLU318:OE1 | Negative |
| 8 | A:ARG183:NH2 - B:GLU318:OE2 | 5.50148 | Electrostatic | Attractive Charge | A:ARG183:NH2 | Positive | B:GLU318:OE2 | Negative |
| 9 | B:ARG199:NH1 - A:ASP294:OD1 | 4.29672 | Electrostatic | Attractive Charge | B:ARG199:NH1 | Positive | A:ASP294:OD1 | Negative |
| 10 | B:LYS316:NZ - A:ASP157:OD1 | 4.91722 | Electrostatic | Attractive Charge | B:LYS316:NZ | Positive | A:ASP157:OD1 | Negative |
| 11 | A:HIS110:HE2 - B:LYS317:O | 2.35866 | Hydrogen Bond | Conventional Hydrogen Bond | A:HIS110:HE2 | H-Donor | B:LYS317:O | H-Acceptor |
| 12 | A:LYS224:HZ2 - B:ILE369:O | 2.41185 | Hydrogen Bond | Conventional Hydrogen Bond | A:LYS224:HZ2 | H-Donor | B:ILE369:O | H-Acceptor |
| 13 | A:LYS224:HZ3 - B:ILE369:O | 2.84512 | Hydrogen Bond | Conventional Hydrogen Bond | A:LYS224:HZ3 | H-Donor | B:ILE369:O | H-Acceptor |
| 14 | A:LYS268:HZ1 - B:ASP364:O | 2.45355 | Hydrogen Bond | Conventional Hydrogen Bond | A:LYS268:HZ1 | H-Donor | B:ASP364:O | H-Acceptor |
| 15 | B:LYS285:HZ3 - A:THR109:OG1 | 2.54351 | Hydrogen Bond | Conventional Hydrogen Bond | B:LYS285:HZ3 | H-Donor | A:THR109:OG1 | H-Acceptor |
| 16 | B:LYS285:HZ3 - A:SER133:OG | 1.89906 | Hydrogen Bond | Conventional Hydrogen Bond | B:LYS285:HZ3 | H-Donor | A:SER133:OG | H-Acceptor |
| 17 | B:ASN347:HD22 - A:ASP131:OD1 | 1.79321 | Hydrogen Bond | Conventional Hydrogen Bond | B:ASN347:HD22 | H-Donor | A:ASP131:OD1 | H-Acceptor |
| 18 | B:GLN368:HE21 - A:ASN245:OD1 | 2.03323 | Hydrogen Bond | Conventional Hydrogen Bond | B:GLN368:HE21 | H-Donor | A:ASN245:OD1 | H-Acceptor |
| 19 | B:GLN368:HE22 - A:ASN245:O | 2.86998 | Hydrogen Bond | Conventional Hydrogen Bond | B:GLN368:HE22 | H-Donor | A:ASN245:O | H-Acceptor |
| 20 | B:LYS370:HZ3 - A:SER178:OG | 1.84334 | Hydrogen Bond | Conventional Hydrogen Bond | B:LYS370:HZ3 | H-Donor | A:SER178:OG | H-Acceptor |
| 21 | A:GLY85:CA - B:SER320:OG | 3.38873 | Hydrogen Bond | Carbon Hydrogen Bond | A:GLY85:CA | H-Donor | B:SER320:OG | H-Acceptor |
| 22 | B:LYS306:CE - A:HIS271:ND1 | 3.28818 | Hydrogen Bond | Carbon Hydrogen Bond | B:LYS306:CE | H-Donor | A:HIS271:ND1 | H-Acceptor |
| 23 | B:LYS370:NZ - A:PHE201 | 4.08798 | Electrostatic | Pi-Cation | B:LYS370:NZ | Positive | A:PHE201 | Pi-Orbitals |
| 24 | B:GLU318:OE1 - A:TYR134 | 4.60367 | Electrostatic | Pi-Anion | B:GLU318:OE1 | Negative | A:TYR134 | Pi-Orbitals |
| 25 | B:GLY319:HN - A:HIS110 | 2.29603 | Hydrogen Bond | Pi-Donor Hydrogen Bond | B:GLY319:HN | H-Donor | A:HIS110 | Pi-Orbitals |
| 26 | B:ALA305:CB - A:HIS271 | 3.56198 | Hydrophobic | Pi-Sigma | B:ALA305:CB | C-H | A:HIS271 | Pi-Orbitals |
| 27 | B:VAL340:CG2 - A:PHE269 | 3.68112 | Hydrophobic | Pi-Sigma | B:VAL340:CG2 | C-H | A:PHE269 | Pi-Orbitals |
| 28 | A:LYS268 - B:LYS338 | 4.28766 | Hydrophobic | Alkyl | A:LYS268 | Alkyl | B:LYS338 | Alkyl |
| 29 | A:PHE80 - B:LYS349 | 4.94473 | Hydrophobic | Pi-Alkyl | A:PHE80 | Pi-Orbitals | B:LYS349 | Alkyl |
| 30 | A:TYR82 - B:VAL322 | 5.03493 | Hydrophobic | Pi-Alkyl | A:TYR82 | Pi-Orbitals | B:VAL322 | Alkyl |
| 31 | A:HIS110 - B:LYS285 | 4.73543 | Hydrophobic | Pi-Alkyl | A:HIS110 | Pi-Orbitals | B:LYS285 | Alkyl |
| 32 | A:PHE201 - B:LYS370 | 4.98742 | Hydrophobic | Pi-Alkyl | A:PHE201 | Pi-Orbitals | B:LYS370 | Alkyl |
| 33 | A:HIS271 - B:LYS306 | 4.68625 | Hydrophobic | Pi-Alkyl | A:HIS271 | Pi-Orbitals | B:LYS306 | Alkyl |
| 34 | A:HIS271 - B:VAL340 | 4.80253 | Hydrophobic | Pi-Alkyl | A:HIS271 | Pi-Orbitals | B:VAL340 | Alkyl |
| S. No | Name | Distance | Category | Type | From | From Chemistry | To | To Chemistry |
|---|---|---|---|---|---|---|---|---|
| 1 | A:LYS225:HZ1 - B:ASP311:OD2 | 1.62127 | Hydrogen Bond; Electrostatic | Salt Bridge; Attractive Charge | A:LYS225:HZ1 | H-Donor; Positive | B:ASP311:OD2 | H-Acceptor; Negative |
| 2 | A:LYS268:HZ1 - B:ASP364:OD1 | 1.6792 | Hydrogen Bond; Electrostatic | Salt Bridge; Attractive Charge | A:LYS268:HZ1 | H-Donor; Positive | B:ASP364:OD1 | H-Acceptor; Negative |
| 3 | B:LYS142:HZ2 - A:ASP294:OD1 | 3.1038 | Hydrogen Bond; Electrostatic | Salt Bridge; Attractive Charge | B:LYS142:HZ2 | H-Donor; Positive | A:ASP294:OD1 | H-Acceptor; Negative |
| 4 | B:LYS285:HZ1 - A:ASP131:OD1 | 1.64033 | Hydrogen Bond; Electrostatic | Salt Bridge; Attractive Charge | B:LYS285:HZ1 | H-Donor; Positive | A:ASP131:OD1 | H-Acceptor; Negative |
| 5 | B:LYS316:HZ3 - A:ASP203:OD2 | 1.56125 | Hydrogen Bond; Electrostatic | Salt Bridge; Attractive Charge | B:LYS316:HZ3 | H-Donor; Positive | A:ASP203:OD2 | H-Acceptor; Negative |
| 6 | B:LYS349:HZ2 - A:ASP56:OD1 | 1.61466 | Hydrogen Bond; Electrostatic | Salt Bridge; Attractive Charge | B:LYS349:HZ2 | H-Donor; Positive | A:ASP56:OD1 | H-Acceptor; Negative |
| 7 | A:ARG183:NH1 - B:GLU318:OE1 | 4.22015 | Electrostatic | Attractive Charge | A:ARG183:NH1 | Positive | B:GLU318:OE1 | Negative |
| 8 | A:ARG183:NH2 - B:GLU318:OE2 | 5.50148 | Electrostatic | Attractive Charge | A:ARG183:NH2 | Positive | B:GLU318:OE2 | Negative |
| 9 | B:ARG199:NH1 - A:ASP294:OD1 | 4.29672 | Electrostatic | Attractive Charge | B:ARG199:NH1 | Positive | A:ASP294:OD1 | Negative |
| 10 | B:LYS316:NZ - A:ASP157:OD1 | 4.91722 | Electrostatic | Attractive Charge | B:LYS316:NZ | Positive | A:ASP157:OD1 | Negative |
| 11 | A:HIS110:HE2 - B:LYS317:O | 2.35866 | Hydrogen Bond | Conventional Hydrogen Bond | A:HIS110:HE2 | H-Donor | B:LYS317:O | H-Acceptor |
| 12 | A:LYS224:HZ2 - B:ILE369:O | 2.41185 | Hydrogen Bond | Conventional Hydrogen Bond | A:LYS224:HZ2 | H-Donor | B:ILE369:O | H-Acceptor |
| 13 | A:LYS224:HZ3 - B:ILE369:O | 2.84512 | Hydrogen Bond | Conventional Hydrogen Bond | A:LYS224:HZ3 | H-Donor | B:ILE369:O | H-Acceptor |
| 14 | A:LYS268:HZ1 - B:ASP364:O | 2.45355 | Hydrogen Bond | Conventional Hydrogen Bond | A:LYS268:HZ1 | H-Donor | B:ASP364:O | H-Acceptor |
| 15 | B:LYS285:HZ3 - A:THR109:OG1 | 2.54351 | Hydrogen Bond | Conventional Hydrogen Bond | B:LYS285:HZ3 | H-Donor | A:THR109:OG1 | H-Acceptor |
| 16 | B:LYS285:HZ3 - A:SER133:OG | 1.89906 | Hydrogen Bond | Conventional Hydrogen Bond | B:LYS285:HZ3 | H-Donor | A:SER133:OG | H-Acceptor |
| 17 | B:ASN347:HD22 - A:ASP131:OD1 | 1.79321 | Hydrogen Bond | Conventional Hydrogen Bond | B:ASN347:HD22 | H-Donor | A:ASP131:OD1 | H-Acceptor |
| 18 | B:GLN368:HE21 - A:ASN245:OD1 | 2.03323 | Hydrogen Bond | Conventional Hydrogen Bond | B:GLN368:HE21 | H-Donor | A:ASN245:OD1 | H-Acceptor |
| 19 | B:GLN368:HE22 - A:ASN245:O | 2.86998 | Hydrogen Bond | Conventional Hydrogen Bond | B:GLN368:HE22 | H-Donor | A:ASN245:O | H-Acceptor |
| 20 | B:LYS370:HZ3 - A:SER178:OG | 1.84334 | Hydrogen Bond | Conventional Hydrogen Bond | B:LYS370:HZ3 | H-Donor | A:SER178:OG | H-Acceptor |
| 21 | A:GLY85:CA - B:SER320:OG | 3.38873 | Hydrogen Bond | Carbon Hydrogen Bond | A:GLY85:CA | H-Donor | B:SER320:OG | H-Acceptor |
| 22 | B:LYS306:CE - A:HIS271:ND1 | 3.28818 | Hydrogen Bond | Carbon Hydrogen Bond | B:LYS306:CE | H-Donor | A:HIS271:ND1 | H-Acceptor |
| 23 | B:LYS370:NZ - A:PHE201 | 4.08798 | Electrostatic | Pi-Cation | B:LYS370:NZ | Positive | A:PHE201 | Pi-Orbitals |
| 24 | B:GLU318:OE1 - A:TYR134 | 4.60367 | Electrostatic | Pi-Anion | B:GLU318:OE1 | Negative | A:TYR134 | Pi-Orbitals |
| 25 | B:GLY319:HN - A:HIS110 | 2.29603 | Hydrogen Bond | Pi-Donor Hydrogen Bond | B:GLY319:HN | H-Donor | A:HIS110 | Pi-Orbitals |
| 26 | B:ALA305:CB - A:HIS271 | 3.56198 | Hydrophobic | Pi-Sigma | B:ALA305:CB | C-H | A:HIS271 | Pi-Orbitals |
| 27 | B:VAL340:CG2 - A:PHE269 | 3.68112 | Hydrophobic | Pi-Sigma | B:VAL340:CG2 | C-H | A:PHE269 | Pi-Orbitals |
| 28 | A:LYS268 - B:LYS338 | 4.28766 | Hydrophobic | Alkyl | A:LYS268 | Alkyl | B:LYS338 | Alkyl |
| 29 | A:PHE80 - B:LYS349 | 4.94473 | Hydrophobic | Pi-Alkyl | A:PHE80 | Pi-Orbitals | B:LYS349 | Alkyl |
| 30 | A:TYR82 - B:VAL322 | 5.03493 | Hydrophobic | Pi-Alkyl | A:TYR82 | Pi-Orbitals | B:VAL322 | Alkyl |
| 31 | A:HIS110 - B:LYS285 | 4.73543 | Hydrophobic | Pi-Alkyl | A:HIS110 | Pi-Orbitals | B:LYS285 | Alkyl |
| 32 | A:PHE201 - B:LYS370 | 4.98742 | Hydrophobic | Pi-Alkyl | A:PHE201 | Pi-Orbitals | B:LYS370 | Alkyl |
| 33 | A:HIS271 - B:LYS306 | 4.68625 | Hydrophobic | Pi-Alkyl | A:HIS271 | Pi-Orbitals | B:LYS306 | Alkyl |
| 34 | A:HIS271 - B:VAL340 | 4.80253 | Hydrophobic | Pi-Alkyl | A:HIS271 | Pi-Orbitals | B:VAL340 | Alkyl |
PGIP, 1BHE, bPGIP-1BHE, PvPGIP2-1BHE and mutated PvPGIP2-1BHE complexes were submitted for MD simulations of 50ns in GROMACS. All systems were solvated in a cubic box. To gauge the intrinsic stability and dynamics of each system, the different stability parameters were calculated from each resultant trajectory. The backbone RMSD analysis provides important information on the stability of protein and protein-protein complexes and the time when simulation reached equlibrium. RMSD profiles of bPGIP and 1BHE displayed least deviation with average RMSD ~0.25nm during the simulation period and by the time of 40 ns all both systems achieved convergence (Figure 4A).
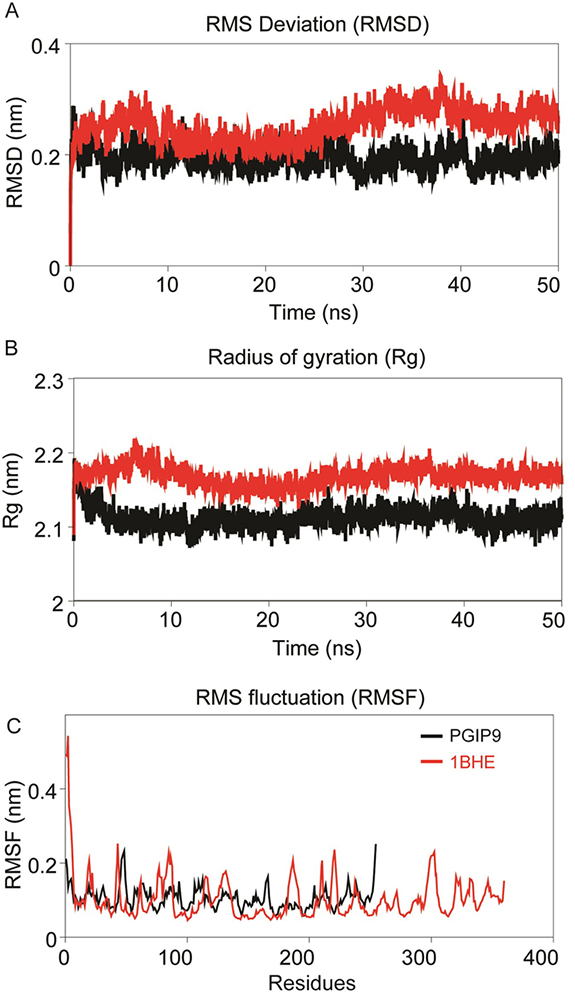 Figure 4
Figure 4
Intrinsic dynamics stabilities of the modeled bPGIP and experimental structure of EcPG (1BHE). (A) Root mean squared deviation (RMSD) of backbone atoms during the MD simulations as function of time. (B) Radius of gyration (Rg, nm) of the structures during the MD simulation is plotted along the trajectory; snapshots were acquired at every 20 picosecond interval. (C) Cα-Root mean squared fluctuation (RMSF, nm) of the structures during 50 ns MD simulation.
The residue flexibility of bPGIP and 1BHE structures was examined by performing Cα RMSF analysis. Both the protein displayed differential flexibility where 1BHE protein gave a maximum value of 0.53 nm and bPGIP with average RMSF value of 0.23nm (Figure 4C). The RMSF values remarkably demonstrate the difference in residual flexibility in both the proteins. Radius of gyration (Rg) represents the atomic distribution from their mutual center of mass in terms of mass-weighted root mean square distance (40-42). The Rg depicts the compactness and inclusive dimension of the protein and protein-protein complexes that may comprise their appropriate interactions. Statistical analysis of Rg for all the systems throughout the 50 ns trajectory showed distinctive difference for all protein (Figure 4B) and protein-protein complexes. Lie the RMSD, 1BHE system had higher Rg value than that of bPGIP system (~2.13 nm) (Figure 4B). RMSD profile of all three complexes bPGIP-1BHE, 1OGQ-1BHE and mutant 1OGQ-1BHE systems displayed least deviation (Figure 5A). In case of protein-protein complexes, bPGIP-1BHE displayed least gyradius indicating its compact packing and stability. The Rg pattern of 1OGQ-1BHE and mutant 1OGQ-1BHE systems followed a constant trend in radius of gyration with a gyradius of 2.83 and 2.84 nm, respectively (Figure 5B). Solvent accessible surface area (SASA) is another significant measure that calculates accessible area of the solvent molecule. SASA was also computed for 50 ns for all the proteins. In bPGIP, total SASA changed from 144.41 nm2 (with 56.95 nm2 as hydrophobic surface area and 87.46 nm2 as hydrophilic surface area) at time t=0 ns to 145.173 nm2 (with 56.46 nm2 as hydrophobic surface area and 88.71 nm2 as hydrophilic surface area) at time t=50 ns. For 1BHE, total SASA shifted from 175.28 nm2 (with 52.79 nm2 as hydrophobic surface area and 122.50 nm2 as hydrophilic surface area) at time t=0 ns to 181.704 nm2 (with 52.3676 nm2 as hydrophobic surface area and 129.336 nm2 as hydrophilic surface area) at 50 ns. SASA in complex (bPGIP-1BHE) drifted from 306.86 nm2 (with 105.12 nm2 as hydrophobic surface area and 201.738 nm2 as hydrophilic surface area) at time 0 ns to 315.59 nm2 (with 106.94 nm2 as hydrophobic surface area and 208.65 nm2 as hydrophilic surface area) at time 50 ns. Our study displayed 11.289 nm2 surface area was interacting between bPGIP and 1BHE thus leading to total 14 bonds formation. The SASA was computed for complex PvPGIP2-1BHE changed from 165.73 nm2 (with 88.32 nm2 as hydrophobic surface area and 77.41 nm2 as hydrophilic surface area) at time t=0 ns to 159.61 nm2 (with 83.45 nm2 as hydrophobic surface area and 76.16 nm2 as hydrophilic surface area) at time t=50 ns. In the same way, SASA was also computed for mutated complex (PvPGIP2-1BHE) changed from 163.54 nm2 (with 87.19 nm2 as hydrophobic surface area and 76.35 nm2 as hydrophilic surface area) at time t=0 ns to 158.92 nm2 (with 80.71 nm2 as hydrophobic surface area and 78.21 nm2 as hydrophilic surface area) at time t=50 ns. We also computed the evolution of secondary structure elements from the resultant trajectories of bPGIP and EcPG using DSSP algorithm, which displayed subtle variation of secondary structure elements including turns ad the β-strands during MD (Figure 3,6). However, the structural fold of the both the PGP and PGIP were found to be intact.
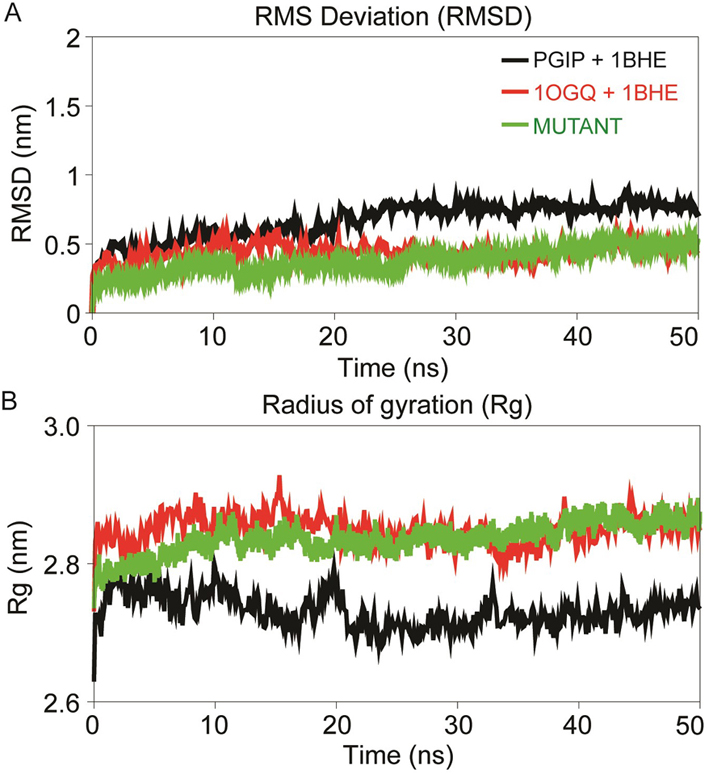 Figure 5
Figure 5
Dynamics stability profile displaying the RMSD of backbone atoms (A) and radius of gyration (Rg) profile of the PG-PGIP complexes over the time scale of 50 ns MD (B). The black curve of PGIP+1BHE represent the bPGIP with EcPG complex, the red curve of 1OGQ+1BHE represents the PvPGIP2 with EcPG and green curve of mutant represents the mutated mPvPGIP2 with EcPG.
The intermolecular hydrogen bonds (H-bonds) between interacting atom pairs in a protein-protein complex plays vital role in stability and molecular recognition processs (Dehury et al., 2014). To gauge the dynamics stability of each complex i.e., bPGIP-1BHE, 1OGQ(PvPGIP2)-1BHE and mutated PvPGIP2-1BHE, the intermolecular H-bonds were calculated with respect to time during the 50 ns MD simulations. All the complexes displayed a pattern of differential H-bonding with additional number of H-bonds (Figure 7).
 Figure 6
Figure 6
Evolution of secondary structure elements of bPGIP and EcPG during 50 ns MD simulation. DSSP was employed to calculate the secondary structure elements of PG and PGIP during MD simulation.
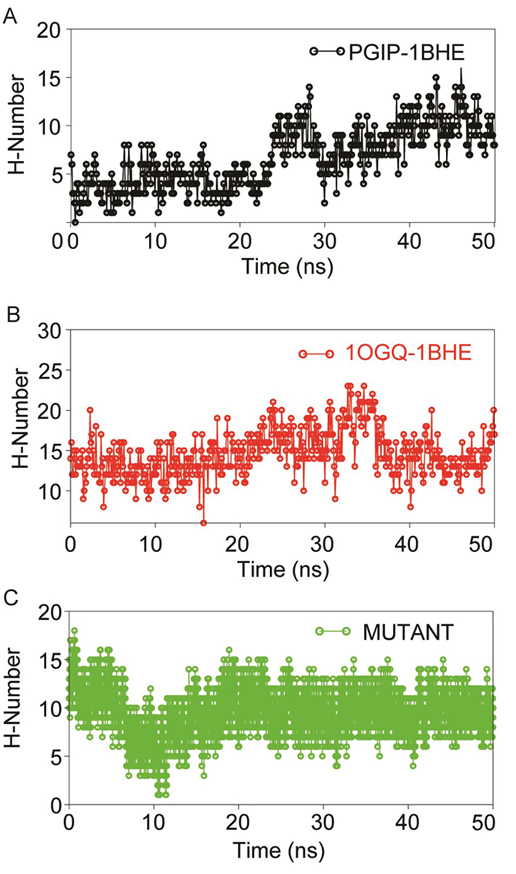 Figure 7
Figure 7
Dynamics stability of protein-protein complexes (A) PGIP-1BHE (B) 1OGQ-1BHE) and (C) mutant 1OGQ-1BHE inferred through inter-molecular H-bond analyses using gmx hbond utility of GROMACS over 50 ns MD in aqueous solution.
In all systems during initial 20 ns there was significant decrease in H-bonds were observed which later increased and remained stable till 50 ns. Though, in all complexes, some of the imperative H-bonds were broken out during MD simulation, but at later stage they well remunerated by new H-bonds, van der Waals and hydrophobic contacts. This may be due to structural re-orientation of interacting proteins within the binding pocket. The change in intermolecular H-bonds and hydrophobic contacts detected before and after MD in bPGIP-1BHE(EcPG) complexes perfectly compare with the results of PCA (as discussed later). The bPGIP-1BHE (Figure 7A) system displayed least (Average ~11.23) number of H-bonds followed 1OGQ-1BHE (Figure 7B) system with ~15.23 number of H-bonds. As compared to the other two systems, the mutant PvPGIP2-1BHE system displayed a stable H-bonding pattern with an average of ~13.98 number of H-bonds (Figure 7C).
The activity of bPGIP and 1BHE was interrelated with the protein motion through essential dynamics calculations bPGIP and 1BHE interaction significantly influences the whole protein motion and further their function. Structural reorganizations are imperative for appropriate signaling. Dynamic cross-correlation was performed to study the motion of residues (data not shown). The correlation value lies between -0.0193 (blue) to 0.104 (red) for bPGIP and -0.00977 (blue) and 0.0379 (red) for 1BHE which signifies negative and positive correlation correspondingly. Covariance analysis expounds the positive and negative correlated motions in the protein. A noteworthy alteration was observed between the covariance of motion among all the complexes. Overall, the bPGIP-1BHE displayed increased collective motion, while decreasing the collective motion was observed in the 1OGQ(PvPGIP2)-1BHE and mutant PvPGIP2-1BHE complexes. Both ways, the conformational and flexibility changes give the impression to play a crucial role in bPGIP-ecPG interactions. For PCA analysis, we only restricted the first ten and/or twenty modes for analysis of the essential subspace as they explain >87% variance of two systems (Figure 8A, B)
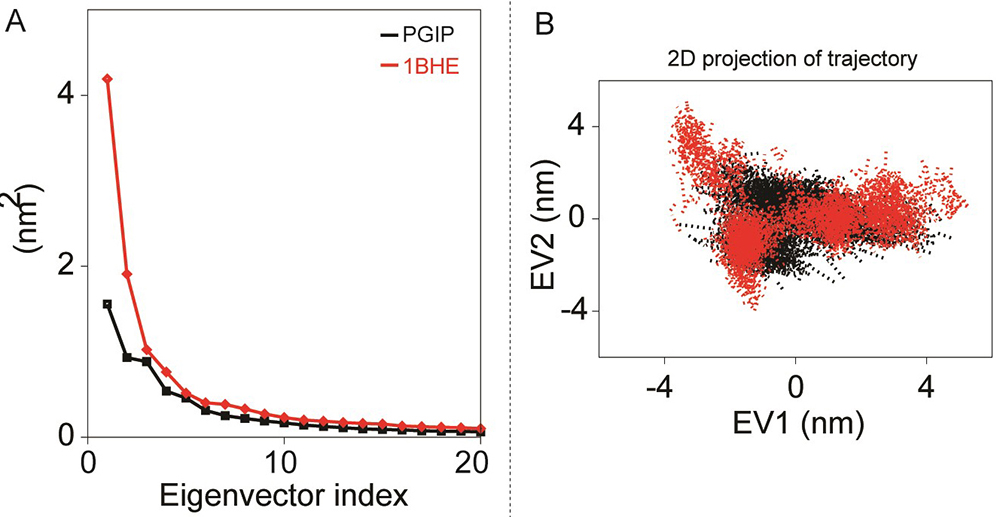 Figure 8
Figure 8
Principal component analyses of bPGIP and EcPG during 50 ns all-atoms MD simulation. (A) The eigenvalues for the bPGIP and EcPG models as a function of eigenvector. The main plot displays the eigenvalues of only the first 20 eigenvectors. (B) The cloud represents the 50 ns trajectories of bPGIP and EcPG models projected onto the first two eigenvectors.
In this study we only considered the first two principal components (i.e., PC1 and PC2) which govern the fluctuations of protein conformation. By projecting the trajectory on specific vector, the change of a trajectory alongside each eigenvector can be detected. A greater dispersal of dots specifies more variance congruous with the higher changes in conformations in bPGIP-1BHE complex (Figure 9A). In case of proteins, 1BHE system occupied a large conformation sub-space while bPGIP with least sub-space with a trace of covariance of 15.3477 and 8.7281 nm2. Protein trajectories analysis exposed a least subspace dimension and decreased concerted motions in 1OGQ (PvPGIP2)-1BHE complex and mutant PvPGIP2-1BHE complex with a trace of covariance of 25.78 and 15.3914 nm2 respectively. Then bPGIP-1BHE system covered a larger subspace (with a trace value 37.7326 nm2) with exceptionally higher variation. As exposed by the trajectory projection along EV1 and EV2, bPGIP-1BHE exhibited a bigger value of trace of covariance matrix than the other two complexes (Figure 9B).
 Figure 9
Figure 9
PCA of the PGIP-PG models. (A). Eigenvalues for the PGIP-PG complex models as a function of eigenvector (B). Projection of top two eigenvectors (EV1 and EV2) into the phase space.
Clustering approach was applied using the gmx cluster toolkit to explore the conformation heterogeneity of the resulted protein structures from the MD simulations. For clustering analysis, the GROMOS clustering algorithm was employed with a Cα-RMSD cutoff of 0.25 nm. In case of bPGIP-1BHE complex clusters, the RMSD was observed within the range of 0.0609 to 0.601 nm (with average RMSD of 0.321875 nm). A total of 17 dominant clusters were obtained consisting of 93% of the total protein structures for bPGIP-1BHE complex indicating structural conformational changes between these proteins. While 10 dominant clusters consisting ~89 % of the total protein structures was captured in case of 1OGQ(PvPGIP2)-1BHE complex system. While in case of mutant PvPGIP2-1BHE complex, we found only three dominant clusters ~83 % of total protein structures which indicates least conformational change upon binding. The RMSD was within the range of 0.0633 to 0.455 nm with average of 0.226042 nm. Protein-protein complex frames from the resultant MD trajectory were extracted using cluster analysis technique have been displayed in Figure 10.
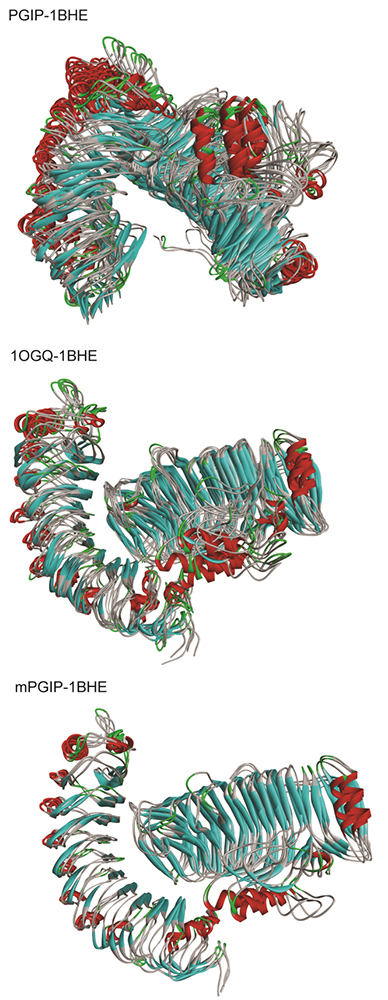 Figure 10
Figure 10
Clustering analyses of the PGIP-PG complexes. The structural superimposition of the top ranked clusters obtained from clustering using GROMOS algorithm with a RMSD cut-off of 0.25 nm (The left structure shows the PGIP while the right structure displays the EcPG).
To understand the molecular recognition of PGIPs to PGs, the interatomic dynamic distances between the interacting atom pairs were calculated from the resultant trajectories for each PGIP-PG complexes (as shown in Figure 11).
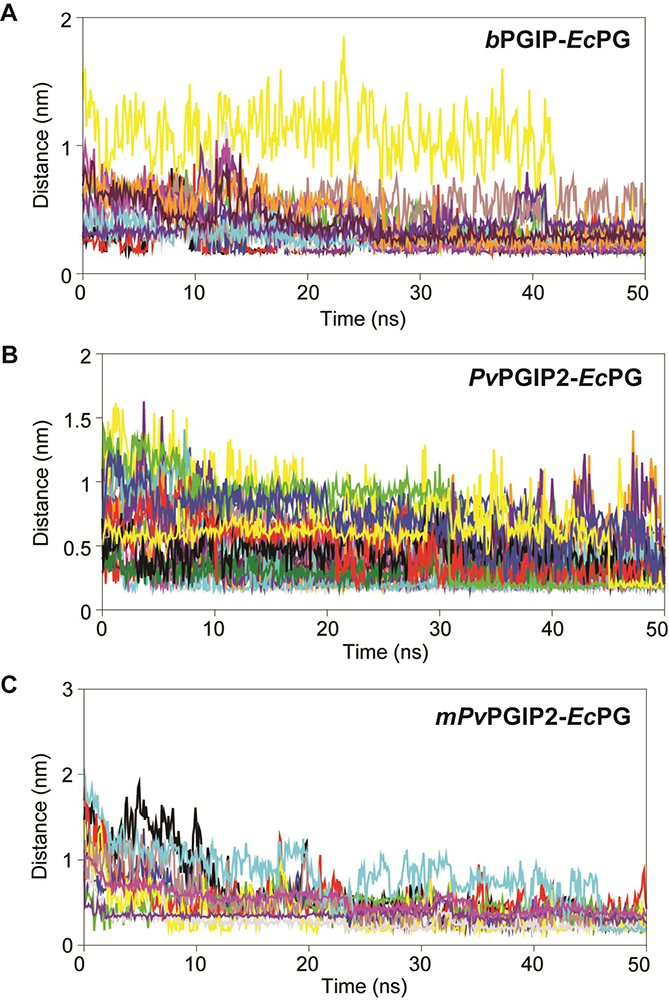 Figure 11
Figure 11
Inter-atomic distance profile of important interacting pair’s (A) bPGIP-EcPG (B) PvPGIP2-EcPG (C) mPvPGIP2-EcPG forming strong hydrogen bonds and electrostatic contacts over the 50 ns trajectory.
The inter-atomic distance of the important electrostatic intercations and hydrogen bond forming pairs were calculated from the 50 ns trajectory. In case of bPGIP-1BHE complex, one salt bridge was noticed between OD1 atom of Asp47 (bPGIP) and CA of Asn303 (1BHE) with an atomic distance of 2.11 Å. Furthermore, a total of three electrostatic contacts were also noticed between Lys20, Trp264 (of bPGIP) with Glu336 and Arg4 of 1BHE. Moreover, three strong H-bonds were noticed between Asp147 (bPGIP) with Asn303 (1BHE) with an average atomic distance 2.7 Å. The other important interacting atom pairs between bPGIP and 1BHE system along with their chemistry has been summarized in Table 4. Furthermore, the inter-atomic distance between interacting atom pairs forming electrostatic and H-bonds in bPGIP and 1BHE has been calculated over 50 ns time (as shown in Figure 11A). Unlike, bPGIP-1BHE system, more number of H-bonds, salt bridges and hydrophobic contacts were observed in 1OGQ(PvPGIP2)-1BHE system (Figure 11B and Table 4).
| Interacting pairs | Distance | Type | Category |
|---|---|---|---|
| A. Interaction analysis of bPGIP with EcPG bPGIP2 | |||
| B:LYS302:HZ3 - A:ASP151:OD1 | 2.1133 | Hydrogen Bond; Electrostatic | Salt Bridge; Attractive Charge |
| A:LYS98:NZ - B:GLU336:OE1 | 5.5707 | Electrostatic | Attractive Charge |
| A:TYR99:HH - B:GLU336:O | 1.6465 | Hydrogen Bond | Conventional Hydrogen Bond |
| A:TRP104:HE1 - B:ASN303:OD1 | 2.4555 | Hydrogen Bond | Conventional Hydrogen Bond |
| A:SER122:HG - B:GLU336:OE1 | 1.6625 | Hydrogen Bond | Conventional Hydrogen Bond |
| B:LYS302:HZ1 - A:HIS149:ND1 | 1.8421 | Hydrogen Bond | Conventional Hydrogen Bond |
| B:ASN303:HD21 - A:ASP125:OD1 | 1.9371 | Hydrogen Bond | Conventional Hydrogen Bond |
| B:ASN303:HD22 - A:ASP125:OD2 | 2.7272 | Hydrogen Bond | Conventional Hydrogen Bond |
| B:SER335:HG - A:THR171:OG1 | 1.8272 | Hydrogen Bond | Conventional Hydrogen Bond |
| B:ASN303:CA - A:ASP125:OD1 | 3.5282 | Hydrogen Bond | Carbon Hydrogen Bond |
| B:ARG4:NH2 - A:TRP242 | 3.5559 | Electrostatic | Pi-Cation |
| B:ARG4:NH2 - A:TRP242 | 3.3407 | Electrostatic | Pi-Cation |
| B:LYS302:HZ2 - A:PHE174 | 2.73 | Hydrogen Bond; Electrostatic | Pi-Cation;Pi-Donor Hydrogen Bond |
| B:ALA305 - A:ARG78 | 4.6706 | Hydrophobic | Alkyl |
| A:PHE54 - B:ALA305 | 4.5288 | Hydrophobic | Pi-Alkyl |
| A:HIS149 - B:LYS302 | 4.3725 | Hydrophobic | Pi-Alkyl |
| B. Interaction analysis of PvPGIP2 with EcPG | |||
| B:LYS285:HZ1 - A:ASP131:OD2 | 2.43337 | Hydrogen Bond; Electrostatic | Salt Bridge; Attractive Charge |
| B:LYS316:HZ2 - A:ASP157:OD1 | 3.08964 | Hydrogen Bond; Electrostatic | Salt Bridge; Attractive Charge |
| A:LYS225:NZ - B:ASP311:OD1 | 4.19795 | Electrostatic | Attractive Charge |
| B:LYS316:NZ - A:ASP131:OD2 | 5.31741 | Electrostatic | Attractive Charge |
| B:LYS350:NZ - A:ASP46:OD1 | 3.2514 | Electrostatic | Attractive Charge |
| A:TYR105:HH - B:ALA348:O | 1.68107 | Hydrogen Bond | Conventional Hydrogen Bond |
| A:TYR107:HH - B:ASN347:O | 1.92245 | Hydrogen Bond | Conventional Hydrogen Bond |
| A:HIS110:HE2 - B:GLU318:OE2 | 1.69691 | Hydrogen Bond | Conventional Hydrogen Bond |
| A:TYR134:HH - B:LYS316:O | 1.98305 | Hydrogen Bond | Conventional Hydrogen Bond |
| A:SER178:HG - B:GLU346:OE2 | 1.59931 | Hydrogen Bond | Conventional Hydrogen Bond |
| A:ARG183:HH12 - B:GLU315:O | 2.43313 | Hydrogen Bond | Conventional Hydrogen Bond |
| A:ARG183:HH22 - B:GLU315:O | 1.7687 | Hydrogen Bond | Conventional Hydrogen Bond |
| A:ASN247:HD22 - B:GLN368:OE1 | 1.81821 | Hydrogen Bond | Conventional Hydrogen Bond |
| A:ASN289:HD22 - B:ASN303:O | 1.69293 | Hydrogen Bond | Conventional Hydrogen Bond |
| B:LYS285:HZ2 - A:SER133:OG | 3.04383 | Hydrogen Bond | Conventional Hydrogen Bond |
| B:LYS306:HZ1 - A:PHE269:O | 2.19959 | Hydrogen Bond | Conventional Hydrogen Bond |
| B:LYS316:HZ3 - A:THR155:OG1 | 2.09442 | Hydrogen Bond | Conventional Hydrogen Bond |
| B:ASN321:HD22 - A:THR109:OG1 | 1.97537 | Hydrogen Bond | Conventional Hydrogen Bond |
| B:LYS349:HZ2 - A:TYR107:OH | 2.92182 | Hydrogen Bond | Conventional Hydrogen Bond |
| B:GLN368:HE22 - A:ASN247:OD1 | 1.83861 | Hydrogen Bond | Conventional Hydrogen Bond |
| B:LYS370:CE - A:THR177:OG1 | 3.73402 | Hydrogen Bond | Carbon Hydrogen Bond |
| B:LYS370:CE - A:THR177:O | 3.77902 | Hydrogen Bond | Carbon Hydrogen Bond |
| B:LYS306:NZ - A:PHE269 | 4.02825 | Electrostatic | Pi-Cation |
| B:GLU318:OE1 - A:TYR134 | 4.47384 | Electrostatic | Pi-Anion |
| B:GLU346:OE1 - A:PHE201 | 3.6823 | Electrostatic | Pi-Anion |
| B:LYS317:C,O;GLU318:N - A:TYR134 | 4.96837 | Hydrophobic | Amide-Pi Stacked |
| A:ALA200 - B:LYS370 | 4.94599 | Hydrophobic | Alkyl |
| A:PHE80 - B:LYS349 | 4.43579 | Hydrophobic | Pi-Alkyl |
| A:PHE269 - B:LYS306 | 4.49725 | Hydrophobic | Pi-Alkyl |
| A:PHE269 - B:VAL340 | 5.06138 | Hydrophobic | Pi-Alkyl |
| C. Interaction analysis of mutant mPvPGIP2 with EcPG | |||
| B:LYS285:HZ1 - A:ASP131:OD2 | 2.43337 | Hydrogen Bond; Electrostatic | Salt Bridge; Attractive Charge |
| B:LYS316:HZ2 - A:ASP157:OD1 | 3.08964 | Hydrogen Bond; Electrostatic | Salt Bridge; Attractive Charge |
| A:LYS225:NZ - B:ASP311:OD1 | 4.19795 | Electrostatic | Attractive Charge |
| B:LYS316:NZ - A:ASP131:OD2 | 5.31741 | Electrostatic | Attractive Charge |
| B:LYS350:NZ - A:ASP46:OD1 | 3.2514 | Electrostatic | Attractive Charge |
| A:TYR105:HH - B:ALA348:O | 1.68107 | Hydrogen Bond | Conventional Hydrogen Bond |
| A:TYR107:HH - B:ASN347:O | 1.92245 | Hydrogen Bond | Conventional Hydrogen Bond |
| A:HIS110:HE2 - B:GLU318:OE2 | 1.69691 | Hydrogen Bond | Conventional Hydrogen Bond |
| A:TYR134:HH - B:LYS316:O | 1.98305 | Hydrogen Bond | Conventional Hydrogen Bond |
| A:SER178:HG - B:GLU346:OE2 | 1.59931 | Hydrogen Bond | Conventional Hydrogen Bond |
| A:ARG183:HH12 - B:GLU315:O | 2.43313 | Hydrogen Bond | Conventional Hydrogen Bond |
| A:ARG183:HH22 - B:GLU315:O | 1.7687 | Hydrogen Bond | Conventional Hydrogen Bond |
| A:ASN247:HD22 - B:GLN368:OE1 | 1.81821 | Hydrogen Bond | Conventional Hydrogen Bond |
| A:ASN289:HD22 - B:ASN303:O | 1.69293 | Hydrogen Bond | Conventional Hydrogen Bond |
| B:LYS285:HZ2 - A:SER133:OG | 3.04383 | Hydrogen Bond | Conventional Hydrogen Bond |
| B:LYS306:HZ1 - A:PHE269:O | 2.19959 | Hydrogen Bond | Conventional Hydrogen Bond |
| B:LYS316:HZ3 - A:THR155:OG1 | 2.09442 | Hydrogen Bond | Conventional Hydrogen Bond |
| B:ASN321:HD22 - A:THR109:OG1 | 1.97537 | Hydrogen Bond | Conventional Hydrogen Bond |
| B:LYS349:HZ2 - A:TYR107:OH | 2.92182 | Hydrogen Bond | Conventional Hydrogen Bond |
| B:GLN368:HE22 - A:ASN247:OD1 | 1.83861 | Hydrogen Bond | Conventional Hydrogen Bond |
| B:LYS370:CE - A:THR177:OG1 | 3.73402 | Hydrogen Bond | Carbon Hydrogen Bond |
| B:LYS370:CE - A:THR177:O | 3.77902 | Hydrogen Bond | Carbon Hydrogen Bond |
| B:LYS306:NZ - A:PHE269 | 4.02825 | Electrostatic | Pi-Cation |
| B:GLU318:OE1 - A:TYR134 | 4.47384 | Electrostatic | Pi-Anion |
| B:GLU346:OE1 - A:PHE201 | 3.6823 | Electrostatic | Pi-Anion |
| B:LYS317:C,O;GLU318:N - A:TYR134 | 4.96837 | Hydrophobic | Amide-Pi Stacked |
| A:ALA200 - B:LYS370 | 4.94599 | Hydrophobic | Alkyl |
| A:PHE80 - B:LYS349 | 4.43579 | Hydrophobic | Pi-Alkyl |
| A:PHE269 - B:LYS306 | 4.49725 | Hydrophobic | Pi-Alkyl |
| A:PHE269 - B:VAL340 | 5.06138 | Hydrophobic | Pi-Alkyl |
A total of two slat bridges were formed between Asp160:OD2 (1OGQ) with Lys285:HZ1 (1BHE) and Asp186:OD1 with Lys316:HZ2 atom pairs respectively. In addition to slat bridges, a total of 15 numbers of H-bonds were also noticed along with three electrostatic contacts (as summarized in Table 4). The HH12 atom of Arg212 (1OGQ) formed two H-bonds with oxygen atom of Glu315 (1BHE) with an atomic distance of 2.43 and 1.76 Å respectively. Similarly, HD22 and OD1 (Asn276:1OGQ) formed two H-bonds with OE1 and HE22 of Gln368 (1BHE). Among the hydrophobic contacts, pi-cation, pi-anion, amide-pi stacked and pi-alkyl interactions were found to dominate the PG-PGIP interactions. In case of mutated PvPGIP2-1BHE complex, a total of four salt bridges were noticed during MD simulation with an average atomic distance of 1.68 Å (i.e., Arg212:HH12-Glu318:OE1, Arg212:HH22-Glu318:OE2, Asp186:OD2-Lys316:HZ1 and Asp232:OD2-Lys316:HZ3 respectively). In addition, four electrostatic contacts, five conventional and two carbon H-bonds were also observed in mutated PvPGIP2-1BHE complex. The inter-atomic distance between corresponding interacting atom pairs of mutated PvPGIP2-1BHE complex has been displayed in Figure 11C. So, from the interatomic distances and in-molecular contact analysis, it can be summarized that the PG-PGIP systems are heterogeneous in nature which is well supported by the MD and PCA analysis. To furtherstrengthen our results, we used the top ranked cluster of each PG-PGIP complexes for electrostatic surface potential calaculation using APBS (Figure 12°- C).
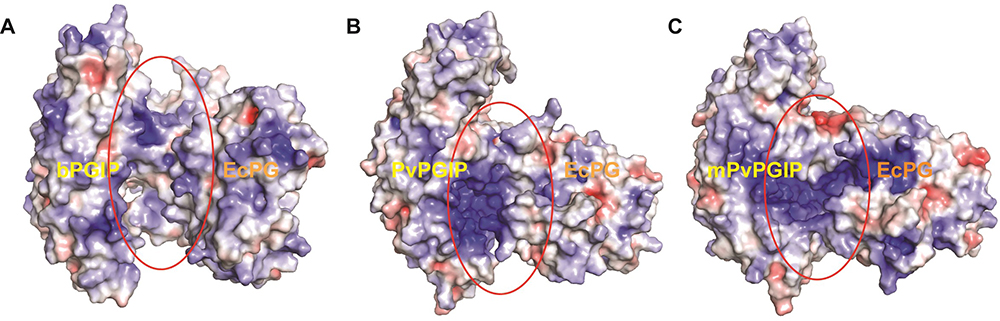 Figure 12
Figure 12
Comparative analyses of electrostatic surface potential of (A) bPGIP-EcPG, (B) PvPGIP2-EcPG and (C) mutant PvPGIP2-EcPG complexes. The structures are representative obtained from the top ranked cluster after clustering analysis.
The mode of binding for PvPGIP2 and mPvPGIP2 to EcPG considerably differs from the mode observed for bPGIP-EcPG. Looking into the electrostatic potential surface for bPGIPs and PvPGIP2, it was evident that a portion of the interacting surface for differs in both where the distribution of positive charged residues dominates in PvPGIP2. The interaction of PvPGIP2 with EcPG dominated by a number of strong H-bonds and eletrostatic contacts, which favors the proper recognition while, it differs significantly in case of bPGIP. The complex PG–PGIP interactions (a model protein–protein interaction system) are said to be of prime importance to understand the plant–pathogen interaction mechanism at atomic scale (Misas-Villamil and Van der Hoorn, 2008). As of now, 3D structure of many PGs have been resolved, however, the only PGIP (i.e., PvPGIP2 of Phaseolus vulgaris) crystal structure has been solved to date. Furthermore, the data existing on the PG–PGIP interactions till date has been based on PvPGIP2. Few previous studies were reported on plant PGIP with fungal PG interactions but as far as our knowledge is concerned not a single study has been reported against the bacterial plant pathogen(s) like Erwinia, Pseudomonas, Pantoea and Ralstonia. Henceforth, to unzip the PG-PGIP interactions i.e., banana PGIP (bPGIP) with Erwinia carotovora PG (EcPG), we employed a combinatorial structural bioinformatics approach including modelling, protein-protein docking and MD simulations to elucidate the plant-pathogen interactions. In silico mutation in Phaseolus vulgaris PGIP (PvPGIP2) was performed and protein-protein docking studies before and after mutation were conducted with ecPG. We performed MD simulations of 50ns on bPGIP, ecPG, bPGIP-ecPG complex, mutated PvPGIP2, and mutated PvPGIP2-EcPG complex. This study will provide a deep understanding of bacterial PG and PGIP interactions in molecular level and inhibitory potential of different PGIPs for EcPG, which may be useful in plant protection paradigm including commercial plantation, vegetable crops and storage conditions.
Various studies have demonstrated that the PGIPs comprised of 10 LRRs in tandem forms a solenoidal shape (43-45). The important motif i.e., LRRs are crucial for PPI in all life forms and diverse cellular processes. Henceforth, it can be suggested that these tandem repeats in fact bear the aforesaid capability to preserve the whole structure of the protein getting in fine alteration in function via selective changes. From our study, it can be observed that the two faces of the solenoid (PGIP) carries very important features where are indispensable for the PG-PGIP interaction. In case of PGIPs, the concave face contributes in establishing the specificity of the plant protein obligatory for binding to the invading PG, while the convex face imparts the required flexibility. This inherent property of defense protein coevolves continuously to fight against the evolving PGs and diversify their recognition capability through the emergence of the multi-gene family to inhibit PGs from different sources (24). Taken together with existing evidences, it can be summarized that the molecular recognition of PG and PGIP interactions are very complex in nature. Structural biology when coupled with site directed mutagenesis can be of immense importance to understand the atomistic interactions of numerous PG–PGIP complexes before a inclusive comprehensive conclusion could be drawn for structure–function association.
Polymorphism of PGIP and PG protein can be used for development of crop protection strategy (46). Site directed mutagenesis of gene PGIP can modulate its function (47). But exploring such variation has major challenges, like existence of less variation in natural population further compounded by methodology of its detection (48). Present work demonstrates that MDS along with site directed mutagenesis has great potential to overcome all such limitations. Such knowledge discovery of desirable alleles of PGIP can be further exploited using genome editing techniques.
Genome editing can improve disease resistance in crop (49). Very recently, genome editing by CRISPR/Cas9 multiplex gene editing strategy has been reported to be very successful in trait modification in banana using Phytoene desaturase (PDS) gene for albinism and dwarfing in Cavendish dessert banana (31) and in Rasthali sweetest Indian banana (50).
The present study reveals that there are few important structural features for PPI which must be deciphered to unravel their functional role. The molecular interaction studies of Musa sp. PGIP with PG molecule from Erwinia carotovora resulted significant insight to understand the relation between resistance and susceptibility of germplasm against PG. The study indicated that specific regions of PGIP are responsible for variation in recognition site to the phytopathogenic Erwinia PG. Similarly, active site of PG from Erwinia carotovora is being intercepted by the PGIP during pathogenesis. PGIP from Musa sp. which is capable of inhibiting Erwinia PG, binds in different and specific mode as compared to PGIPs from other field crops. Electrostatic surface potential observed significant variation in the Musa PGIP interacting surface as compared to other crop plant PGIPs. Hence, it is concluded that electrostatic and van der Waals interactions may play an important role in appropriate recognition of Musa PGIP and Erwinia carotovora PG. This is the novel study of PG-PGIP interaction of any plant pathogenic bacterial PG (Erwinia) using molecular modeling, docking, and MD simulation. Present finding can be of immense use not only as an insight of host-pathogen molecular recognition mechanism but can also be of much pragmatic use in enhancing the banana breeding efficiency against biotic stress obviating environmental and health issues in soft rot disease management in the endeavour of banana productivity.
Authors dully acknowledge to Indian Council of Agricultural Research, New Delhi and Director, ICAR-National Bureau of Agriculturally Important Microorganism, Maunath Bhanjan for providing financial and technical assistance to carry out this research work. The authors also acknowledge the support of computational facilities provided under the Biotechnology Information System Network (BTISNET) grant of DBT-India to the DIC at NBRC, Manesar, India. The authors declare that they have no competing interests. All the authors have read and approved the manuscript. SK, BD and GT conducted the majority of the experiments and performed data analyses. MKS performed part of the molecular dynamic simulation. UBS, YJ, AS, AR and BRP did proof reading of the manuscript. SK designed study and supervised the project. SK, DK, BD, KA, DTN, GT, SJ, MAI and BP wrote the manuscript. SK is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
EcPG
Erwinia carotovora PG
Musa AAB group PGIP
Leucine Rich Repeats
PolyGalacturonase Inhibiting Protein- PolyGalacturonase complex