
Chimeric antigen receptor (CAR) T cell immunotherapy has demonstrated clinical success in treatment of B-cell hematologic cancers. In this study, we compared human Transferrin epitope tagged CAR-T cells with non-tagged CAR-T cells for cytotoxicity, IFN-gamma secretion and tumor clearance in NSG mice. CD19-TF-CAR-T cells had similar cytotoxicity in vitro to CD19-CAR-T cells against cells expressing CD19 antigen: exogenously CD19+ Hela cells and endogenously CD19+ Raji cells. In addition, CD22-TF CAR-T cells were similarly cytotoxic against CD22+ CHO cells and CD22+ Raji cells. Both CD19-TF or CD22-TF-CAR-T cells secreted less IFN-gamma as compared to non-tagged CAR-T cells. In a Raji xenograft NSG mouse model, CD19-TF-CAR-T cells were as effective as CD19-CAR-T cells in reducing tumor growth and extending mouse survival. The results show that CD19-TF-CAR-T cells can be monitored using TF antibody in vitro and ex vivo, and that these cells effectively killed Raji cells in vitro and in vivo with reduced secretion of IFN-gamma. Thus, these TF-tagged CAR-T cells might have improved safety and provide a basis for future clinical studies.
Chimeric antigen receptor (CAR) T cell therapy recently produced excellent results against hematological cancers (1), (2,3), (4), (5). CD19-CAR-T cells had high efficacy against ALL, CML, lymphoma and other types of leukemia (6). Recently two CAR-T cell therapies were approved by FDA: Yescarta and Kymriah (7). Several challenges for CAR-T cell therapy remain to be solved in the future such as the improvement of efficacy against solid tumors and the elimination of cytokine release syndrome (CRS) can encompass high fever, neurological adverse effects, and even mortality (8),(9). Cytokine release syndrome can be managed with tocilizumab (IL-6 pathway inhibitor) (8) but neurotoxicity is still an issue with this approach.
CARs are targeted against tumor-associated antigens (10),(11). The CAR contains an ScFv (single chain variable fragment of antibody targeting tumor antigen) fused to a hinge, transmembrane region, co-stimulatory domain (CD28, 4-1BB, and or GITR (12)) and CD3 zeta activation domain (7), (13,14).
Recently, we demonstrated that Flag epitope tagged CD19 CAR-T cells had equal cytotoxicity to CD19-CAR-T cells and had less cytokine secretion, which could be valuable for the prevention of CRS (15). Flag tag may not be ideal for incorporation into CARs as it is a non-human sequence and therefore potentially immunogenic. We developed a transferrin (TF) antibody specific for a 15 amino-acid peptide and used this peptide tag in our CD19 and CD22 CAR constructs. The CD19-TF- CAR-T cells had similar efficacy as CD19-CAR-T cells both in vitro and in vivo and decreased secretion of IFN-gamma. In addition, CD19-TF performed significantly better than CD19 CAR-T in vivo as measured by better control of tumor growth and longer survival. CD22-TF-CAR-T cells killed Raji and CHO-CD22 cells significantly more than control Mock CAR-T cells and T cells. This data suggest that epitope tagged CAR-T could potentially demonstrate greater efficacy and safety in clinical trial, but must be evaluated on a case-by-case basis.
Hela, Raji and CHO cells were purchased from ATCC. Hela and CHO cells were cultured in DMEM medium (ThermoFisher) with 10% FBS (fetal bovine serum) (Hyclone). Raji were cultured in RPMI-1640 with 10% FBS (ThermoFisher). Both media were supplemented with 1% Penicillin/Streptomycin (ThermoFisher). The stable Hela-CD19+ and CHO-CD22+ cell lines were generated by transduction with lentivirus conferring CD19 and CD22 expression, respectively, and puromycin resistance. Hela-CD19+ and CHO-CD22+ cell lines were maintained in DMEM, 10%FBS and puromycin. HEK293FT cells were provided by Alstem (Richmond, CA) and cultured in DMEM as above.
Genes encoding ScFv from murine anti-human CD19 clone FMC63 (3) or anti-human CD22 clone M971 (16) were synthesized and cloned into a CAR construct with N-terminal GM-CSF signal peptide. Distal to the ScFv were CD28 hinge, transmembrane and costimulatory domains fused to the intracellular domain of CD3 zeta. Two constructs were further modified with the insertion of a 15 amino-acid Transferrin epitope sequence between the ScFv and CD28 hinge. These constructs are herein referred to as CD19-TF-CAR and CD22-TF-CAR. A schema of these constructs is presented in Figure 1. A nonspecific CAR containing ScFv against an intracellular protein was also generated, denoted as mock CAR. CAR genes were subcloned into a third generation lentiviral vector. CAR sequence and orientation were confirmed by Sanger sequencing performed at Quintara Biosciences (South San Francsico, CA).
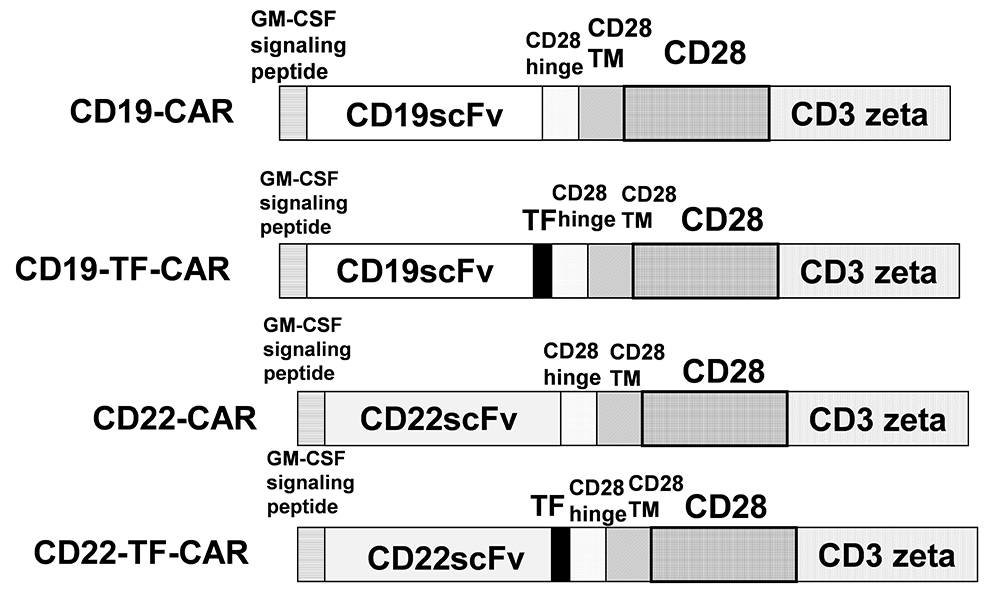 Figure 1
Figure 1
The structure of CD19-TF and CD22-TF- CAR. TF, Transferrin epitope; ScFv, single chain variable fragment. TM is transmembrane domain; CD28 is CD28 co-stimulatory domain; CD3 zeta-activation domain.
VSV-g pseudotyped lentivirus was produced by transfection of HEK293FT with pPACKH1 lentivector packaging mix (System Biosciences, Palo Alto CA) and the CAR vector using Nanofect reagent (Alstem). Supernatants containing lentivirus were harvested at 48-72 hours, centrifuged at 2100g for 30 minutes to remove cellular debris, and ultracentrifuged at 112,000g for 100 minutes to pellet virus. Pellets were resuspended in AIMV medium (ThermoFisher), and aliquots were stored at -800C. Virus was titered after concentration by RT-qPCR using LentiX qRT PCR kit on ABI 7300 thermocycler. Titers were 108-109 vRNA/ml.
Human PBMC (peripheral blood mononuclear cells) were isolated from blood of healthy human donors collected at the Stanford Blood Center according to IRB protocol. The PMBC were purified by Ficoll-paque (GE Health Care) density gradient centrifugation. The cells were cryopreserved in freezing medium (Alstem) and stored in liquid nitrogen until use. PBMC were plated at 1x106 cells/ml in AIMV medium with 10% FCS supplemented with 300U/ml IL-2 in non-treated 24 well plates. T cells were activated with prewashed Human T-cell activator CD3/CD28 Dynabeads (ThermoFisher) at 1:1 bead to cell ratio. At 24 hours, virus (MOI=5) and DEAE dextran at 5 ug/ml were added to the medium. A second transduction with an equal amount of virus was performed at 48 hours. Cultures were expanded over two weeks with AIMV/FCS/IL-2 added as needed to maintain a cell density at 0.5-3 x 106 cells/ml.
A 15-amino acid peptide derived from human transferrin was purchased from Chinese Peptide Company (Hangzhou, China) and conjugated to keyhole limpet hemocyanin. New Zealand white rabbits were injected subcutaneously with the TF-KLH conjugate and complete Freund’s adjuvant (Promab, Changsha, China). Booster injections of TF-KLF were given at 2-3 week intervals to attain sufficient titers to proceed with cells sorting. Rabbit PBMC were isolated from whole blood, and antigen specific B cells isolated by fluorescence activated cell sorting (FACS). RNA was prepared from the cells using the RNeasy Kit (Qiagen) and reverse transcribed. cDNA was used to generate a yeast display library and positive clones were sorted by FACS. Sorted yeast cells were used to create a Pichia expression library. Pichia clones were screened by ELISA for reactivity to TF peptide and full-length transferrin and limited cross-reactivity to several unrelated peptides. A suitably specific TF clone was ultimately generated, and used as anti-TF antibody.
FACS was performed to verify antigen expression on target cells and quantify CAR expression on T cells. 1-5x10^5 cells were transferred to 5ml polystyrene FACS tubes and washed with 4 ml cold FACS buffer (PBS, 0.5%BSA, 2mM EDTA, 0.01% Sodium Azide w/v). Cells were pelleted, decanted and placed on ice. Human serum was added at 1:100, and cells incubated for 10 minutes.
Target cells were incubated with Anti CD19 PE (Clone HIB19 Mouse IgG1, Biolegend, San Diego CA) or Anti CD22 (Clone S-HCL-1 Mouse IgG2b, Biolegend) at 1:20 with 7-aminoactinomycin D at 1:50 (Biolegend). Isotype controls were stained at equivalent antibody concentration (Biolegend).
CAR-T cells expressing the TF tag were stained with anti-TF antibody 1:40 dilution. Cells were stained for 30 minutes on ice and washed once in 4 ml FACS buffer. Cells were then stained with a cocktail consisting of Donkey Anti-Rabbit PE (Jackson Immunoresearch, West Grove PA), CD3 APC (Clone OKT3, Biolegend) and 7-AAD with final dilutions of 1:100, 1:100 and 1:50, respectively.
Non-tagged control CAR-T cells were stained with Goat F(ab2)’ Anti Mouse IgG F(ab2)’ PE (Jackson) at 1:100 for 30 minutes and washed once. Cells were then stained with CD3 APC (1:100) and 7-AAD (1:50).
Blood from retroorbital bleeds was transferred to FACS tubes and resuspended in 4 ml erythrocyte lysis solution (150mM ammonium chloride, 10mM sodium bicarbonate, 1mM EDTA pH 8). White cells were pelleted after 10 minutes and washed 1x in FACS buffer. Cells were stained for CAR-T cells as above.
FACS samples were washed prior to acquisition on BD FACS Calibur (BD). Events were gated on 7AAD negative events and by light scattering for cell sized events.
RTCA was performed on the xCelligence instrument (Acea Biosciences, San Diego CA) with E-Plate 96 in triplicate wells. Adherent cells were plated at 1-4x104 cells/well (Hela and Hela CD19) in 150ul cDMEM and monitored for impedance overnight. Raji cells (suspension) were attached at 1x104 cells/well in E-plates pre-coated with Anti-CD40 (Acea) for 3 hours at room temperature according to the manufacturer provided tethering protocol. At 18-24 hours, CAR-T cells and control Mock CAR-T or T cells were washed to remove IL-2 and resuspended in AIMV/10%FCS. Medium was aspirated from E-plate and effector cells were added at a ratio of 10 or 20 to target cells in 100ul volume. Control wells containing targets cells with medium only were also included. Plates were returned to the instrument and monitored for an additional 24-48 hours.
Raw RTCA impedance data were normalized to the last data point prior to the addition of effector cells. Cytolysis was calculated at the end of the experiment as Ia –IE ____ x100 Ia where Ia is impedance of target cells alone and IE is the impedance of target cells with effectors. Impedance values were normalized to the last data point prior to addition of effector cells and plotted in Prism 7 (Graphpad Software, San Diego CA).
Following RTCA, E-plates were centrifuged at 300g for 5 minutes, and the culture supernatants were collected and frozen. Samples were diluted in PBS/1% BSA (1:5 to 1:10), and detection of IFN-gamma secretion was performed using the Duoset IFN-gamma ELISA kit (R&D Systems, Minneapolis MN) according to manufacturer’s protocol.
All mice experiments were performed according to approved IACUC protocol with six-week-old male NSG mice. Mice were injected intravenously (i.v.) with 5x106 Raji-luciferase cells. On the next day, mice were injected i.v. with 1x107 CAR-T cells or control cells. Bioluminescent imaging to monitor tumor growth was performed with the Xenogen IVIS Spectrum imaging system (PerkinElmer, Waltham WA). Mice were imaged weekly for the first month, and biweekly thereafter. The bioluminescence was measured as total flux in photons/sec and calculated weekly.
For RTCA endpoint cytolysis, cytokine ELISA and bioluminescence data, Student’s t test or one-way ANOVA followed by Tukey test were performed, as described (17). Survival curves were analyzed with log-rank method comparing between each group. A p-value of <0.05 was considered significant.
CD19, CD19-TF, CD22 and CD22-TF CAR lentivirus were used for transduction of T cells (Figure 1). All CAR-T cells were effectively expanded. FACS with TF antibody was performed to detect TF-positive CAR-T cells at day 9 after transduction (Figure 2). T cells transduced with CD19-TF CAR or CD22-TF CAR had increased fluorescence of 1-2 logs compared to non-transduced T cells. After background correction, 54-57% of transduced T cells were positive for the TF epitope showing high level of CAR-positive cells (Figure 2). Thus, TF epitope can be used for detection of TF-positive CD19-TF or CD22-TF-CAR-T cells.
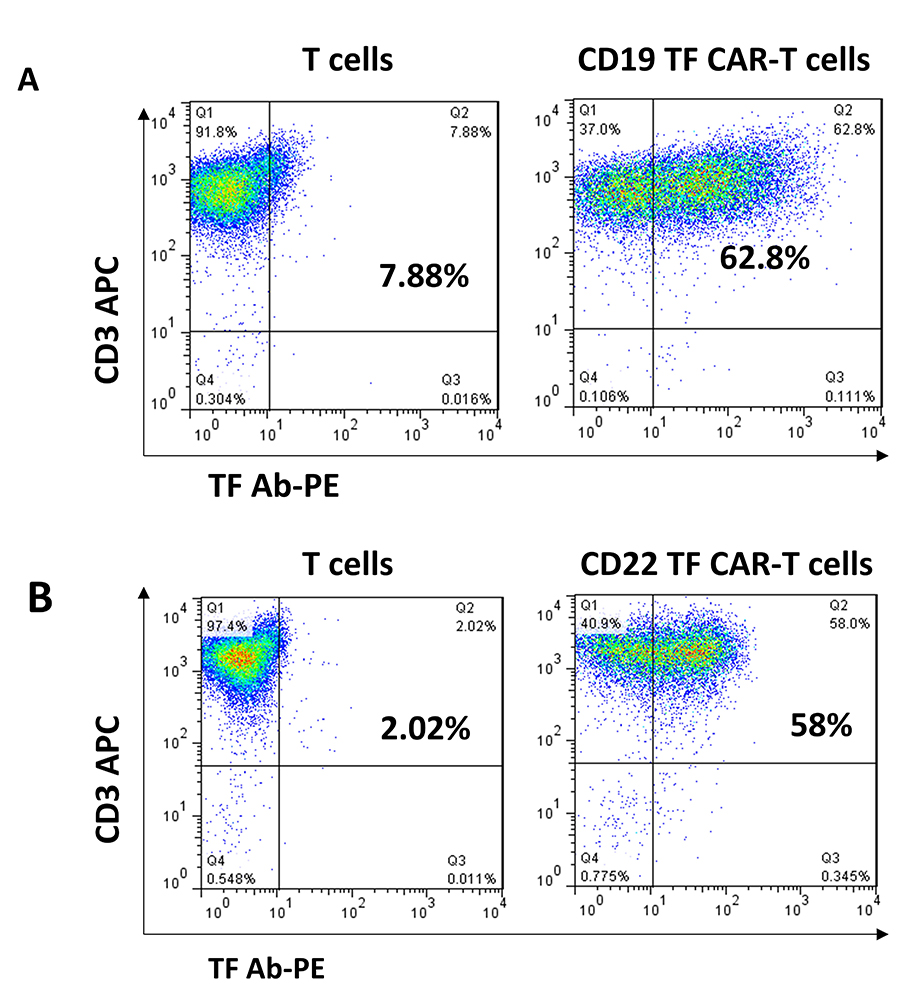 Figure 2
Figure 2
TF antibodies generated against 15 amino-acid TF epitope detect CD19 and CD22-TF- CAR expression. FACS analysis with TF-PE rabbit antibody (X-axis) and CD3-APC antibody (Y-axis) was performed as described in Materials and Methods.
The cytotoxicity of CD19 and CD19-TF CAR-T cells was assayed by RTCA against HeLa cells with and without CD19 expression (Figure 3B-C). Hela cells stably transduced with lentivirus expressing CD19 antigen had high expression of cell surface CD19 detected by FACS with CD19 antibody (Figure 3A). Addition of CD19-TF CAR-T or CD19-CAR-T cells to Hela-CD19 cells caused a significant and nearly complete reduction in the normalized cell index compared with control T cells, Mock-CAR-T cells and CD22-CAR-T cells (Figure 3B). No significant difference in CD19 specific cytotoxicity between CD19-TF and CD19 CAR-T cells was observed, suggesting the TF tag did not interfere with cell killing. Nonspecific killing of Hela (CD19 negative) by CD19-TF and CD19-CAR-T cells was not significantly different from Mock-CAR-T and T cell controls (Figure 3C). Endpoint cytotoxicity of CD19-TF and CD19-CAR-T cells with Hela-CD19 is shown on Figure 3D. There was significantly increased cytolysis of Hela CD19 cells by both CD19-TF and CD19-CAR-T cells compared to mock CAR-T and T cells.
 Figure 3
Figure 3
Real-time cytotoxicity assay (RTCA) with CD19 TF-CAR-T cells, CD19-CAR-T cells and CD19-positive Hela cervical cancer target cells. A. Expression of CD19 in Hela-CD19 stable cell line by FACS with CD19 antibody (black, filled) Isotype control (grey, unfilled). B. RTCA plot with Hela-CD19 target cells. C. RTCA with negative control Hela target cells. 10:1 ratio of Effector to Target cells was used. Mock-CAR-T cells and T cells were used as negative control cells against Hela-CD19-positive cells. Both CD19-TF and CD19-CAR-T cells effectively killed Hela-CD19-positive cells. D. Endpoint cytotoxicity from RTCA. Bars represent % of Cytolysis calculated as described in Materials and Methods. Hela-CD19 ANOVA p < 0.0001, Tukey test * p-value < 0.0001 CD19 or CD19-TF vs mock CAR-T cells.
To examine the effect of the TF tag on CAR specific for another epitope expressed on B cells, we generated CHO cells stably transduced with a high level of CD22 antigen (Figure 4A). CD22-CAR-T cells had a modest but significant increase in cytotoxicity compared to CD22-TF-CAR-T cells against CHO-CD22 (84.8 % ± 0.6 vs 96% ± 0.2%, p = 0.017), and both CARs were significantly more cytotoxic than T cells or Mock-CAR-T cells (Figure 4B, D). None of the effectors tested were cytotoxic against CHO cells without CD22 (Figure 4C-D). Thus, the TF tag did not reduce activity of CD19 CAR and only slightly reduced activity of CD22 CAR.
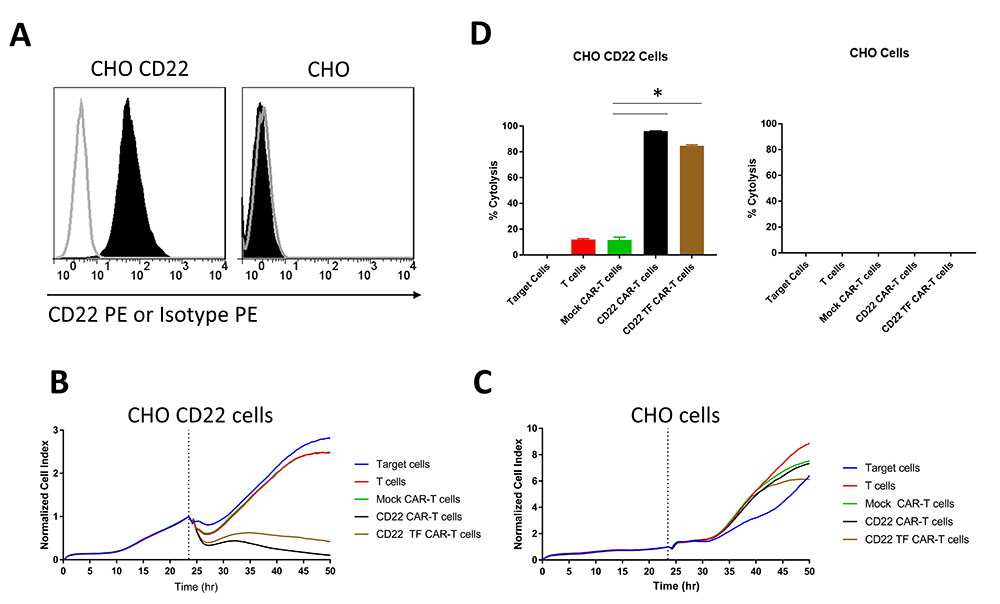 Figure 4
Figure 4
RTCA with CD22 TF-CAR-T cells, CD22-CAR-T cells and CD22-positive CHO target cells. A. Expression of CD22 in CHO-CD22 stable cell line. FACS was performed on CHO-CD22 stable cell line. CD22 antibody (black, filled), Isotype control (grey, unfilled). B. RTCA assay with CD22-TF and CD22-CAR-T cells and CHO-CD22 cells. C. RTCA assay as in B with CHO negative control cells. D. Endpoint Cytotoxicity from RTCA. Bars represent % of Cytolysis calculated as described in Materials and Methods. CHO-CD22 ANOVA p < 0.0001, Tukey test * p-value < 0.0001 CD22 or CD22-TF vs mock CAR-T cells.
Raji cells express CD19 and CD22 and are an appropriate target to assay CD19 and CD22 specific killing. We performed RTCA with CD19-TF and CD22-TF CAR-T cells using Raji cells as targets. Both of the TF tagged CAR-T cells had significantly more Raji killing than mock-CAR-T or T cells (p <0.0001, Figure 5). Similar cytotoxicity was observed with CD19 (15) and CD22-CAR-T cells against Raji cells (Figure 5, right upper panel). These data demonstrate specific and effective killing in a model of endogenous antigen expression.
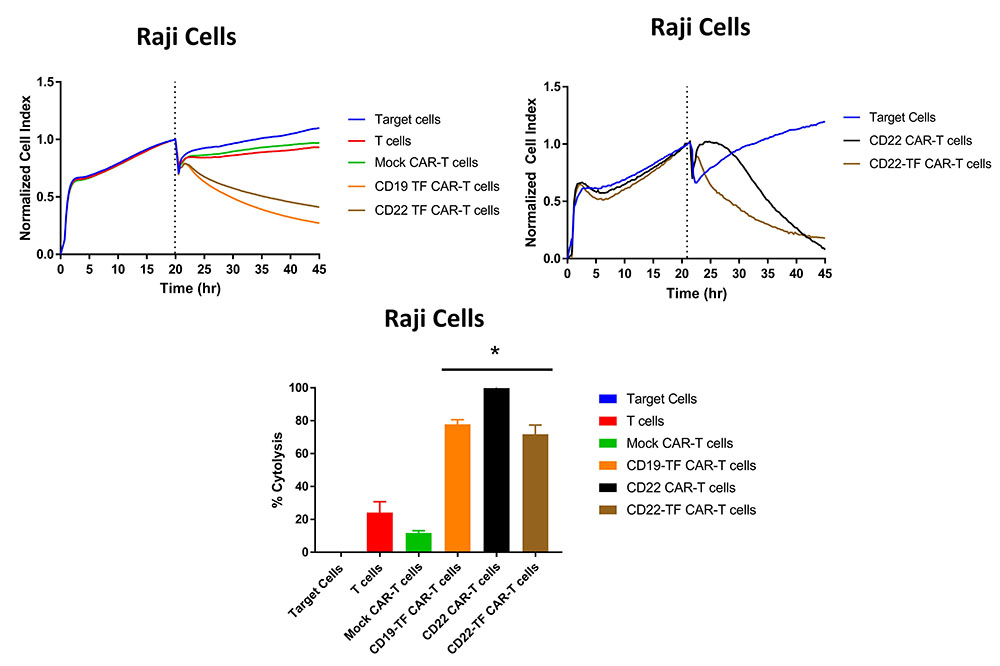 Figure 5
Figure 5
RTCA with Raji cells. Both CD19-TF and CD22-TF effectively killed Raji cells. Left upper panel: RTCA plot; Right upper panel: CD22 and CD22 have similar cytotoxicity against Raji cells. Lower panel: Endpoint cytotoxicity from RTCA. The quantification is based on two independent experiments. Each sample was analyzed in triplicate. ANOVA p < 0.0001, Tukey test * p-value < 0.0001 CD19-TF or CD22-TF vs mock CAR-T cells.
To analyze secretion of IFN-gamma by CAR-T cells, supernatants from Raji cell killing assays were analyzed by ELISA. CD19 CAR-T cells without TF tag had the highest secretion of IFN-gamma (8632 ± 313 pg/ml) (Figure 6A). IFN-gamma secretion by CD19-TF-CAR T cells was nearly 50% lower than CD19-CAR-T cells (4248 ± 202 pg/ml). Similarly, CD22-TF CAR-T secreted significantly lower IFN-gamma than CD22 CAR-T tag against Raji cells (894 ± 23 pg/ml and 1306 ± 13 pg/ml respectively, Figure 5B). Thus, CD19-TF and CD22-CAR-T cells kill Raji cells with lower secretion of IFN-gamma.
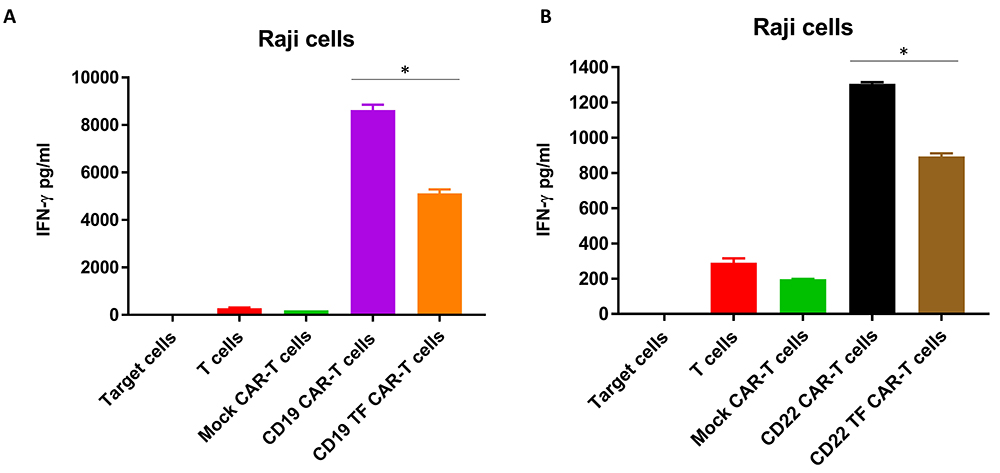 Figure 6
Figure 6
CD19-TF (A) and CD22-TF (B)-CAR-T cells secrete significantly less IFN-gamma than CD19 and CD22-CAR-T cells. A. IFN-gamma secretion from RTCA with CD19-TF CAR-T and CD19 CAR-T (ANOVA p < 0.0001, Tukey test * p < 0.0001 vs CD19-TF vs CD19. B. IFN-gamma secretion from RTCA with CD22-TF CAR-T and CD22 CAR-T (ANOVA p < 0.0001, CD22-TF vs CD22 Tukey test * p < 0.0001).
To test the efficacy of CD19-TF-CAR-T cells in vivo, NSG mice were injected with Raji-Luciferase and, on the following day, CD19-TF CAR-T or CD19 CAR-T or vehicle. Bioluminescent imaging showed a rapid increase in flux in vehicle treated mice between day 7 and 14 (Figure 7A). All vehicle treated mice died by day 18 (median survival 17 days, Figure 7C). CD19 CAR-T treated mice had an increase in flux at day 28, ultimately leading to death in 4 mice by the end of study (median survival 62 days). In the CD19-TF-CAR-T cell group, one mouse had increased flux at day 21 that resolved by day 28. Another mouse in the group died at day 62 without experiencing an increase in tumor flux (median survival >77 days). Both CD19 CAR-T and CD19-TF-CAR-T groups effectively blocked Raji tumor growth and had significantly better survival than vehicle (p = 0.0042 and 0.0025 respectively) but not each other (p = 0.06).
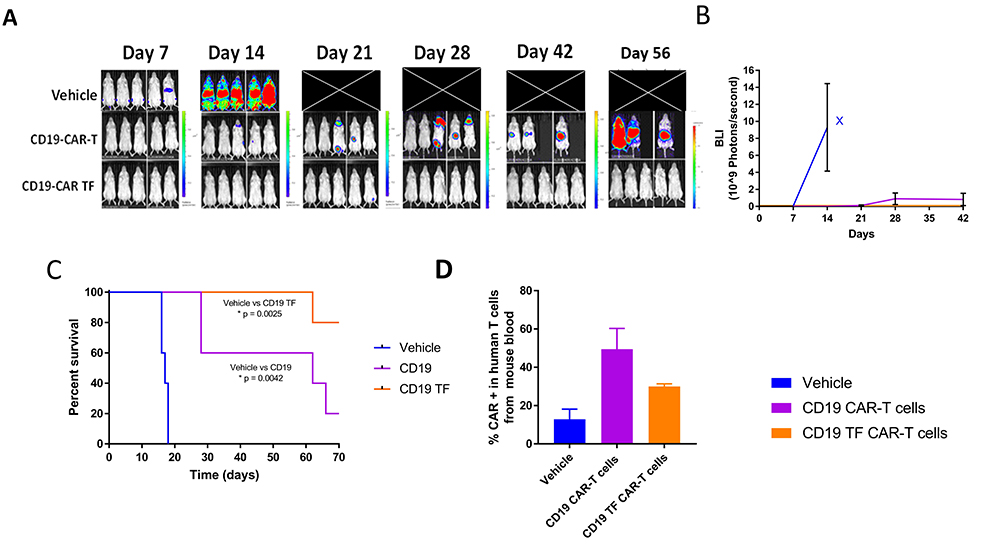 Figure 7
Figure 7
CD19-TF-CAR-T cells significantly decrease Raji xenograft tumor growth. A. In vivo imaging of Raji-luciferase positive xenograft tumors. B. Total Raji-luciferase photon flux as measure of tumor growth (Student’s T-test CD19-TF versus PBS control p < 0.05). C. Kaplan-Meier plot shows significantly increased survival of CD19-TF and CD19-CAR-T cell treated mice versus vehicle treated mice (Log-rank test p <0.0025, p < 0.0042 respectively). D. Detection of CD19-CAR-T cells in mouse blood by FACS Student’s T test CD19-TF CAR-T vs vehicle control p <0.05).
In this study, we evaluated the function of two CAR-T cell constructs that contained the human-derived TF epitope tag. We showed that the TF tag was expressed on the CAR-T cells as detected by anti-TF rabbit antibody. CD19-TF and CD22-TF-CAR cells had comparable cytotoxicity to parental CD19 and CD22-CAR-T cells against Hela-CD19 and CHO-CD22 target cells in vitro. This was also observed against Raji cells endogenously expressing CD19 and CD22 antigens. Moreover, secretion of IFN-gamma by CD19-TF and CD22-TF-CAR-T cells was significantly less that with CD19 and CD22-CAR-T cells. In the in vivo Raji xenograft mouse model, CD19-TF-CAR-T cells performed as well as or better than CD19-CAR-T cells in terms of reduced tumor growth kinetics and overall survival.
TF tagged CAR-T had reduced IFN-gamma secretion compared to non-tagged CAR-T. This parallels our previous findings with FLAG epitope tagged CAR-T. A reduction in cytokine secretion by CAR-T may be desirable in the clinic, as CAR-T therapy frequently causes cytokine release syndrome. The syndrome is characterized by a range of symptoms ranging from fever and nausea to organ dysfunction, hypoxia and life-threatening hypotension. Studies have demonstrated that macrophages play a major role in CRS but have not excluded CAR-T cell derived cytokines. IL-6 is a major mediator of CRS attributed to macrophages (18), (19). We observed less IL-6 secretion by CD19-TF-CAR-T cells than by CD19-CAR-T cells in Raji cells (data not shown), which might be beneficial in reducing severity and occurrence of CRS.
Immunogenicity is a concern in any cell-based therapy. Previously, we reported that CD19-Flag-CAR-T cells were effective in vitro against B cell lymphomas and secreted less cytokines than CD19-CAR-T (15). While the FLAG peptide is not a human sequence, the TF tag is fully human. Thus, the TF tag may be better tolerated leading to more effective CAR-T than FLAG in clinic.
We have also tested the effect of a truncated TF tag and 2 x repeat TF tag to check if the tag length had an effect on cytokine secretion. CAR-T with either tag had the same cytotoxicity against Hela-CD19 cells and Raji as the 1x TF tag (not shown). The truncated TF tag had further decreased IFN-gamma in Raji cell RTCA compared to the 1x TF-tag; however it was not detected by FACS with our TF antibody and is therefore insufficient for assessing transduction efficiency. Cytokine secretion was equivalent between the 1x and 2x TF tagged constructs suggesting no functional advantage to additional tag length beyond 1x. We can explain decreased secretion of IFN-gamma or other cytokines as was shown with Flag tag (15) by increased distance from ScFv to the intracellular signaling co-stimulatory CD28 domain and CD3 activation domain of CAR, and also by changed conformation and changed flexibility of extracellular portion of CAR construct. Interestingly, our previously published data with Flag-tagged CD19-CAR-T cells with reduced cytokines and same cytotoxicity (15), and present report with TF-tagged CD19 and CD22-CAR-T cells were recently confirmed by other study (20). The authors generated by tertiary-structure prediction Phrye2 software a new CD19-41BB-CD3 CAR construct with 10 extra amino-acids of CD8 alpha sequence in the extracellular CD8 hinge region, and 4 extra amino-acids intracellularly and demonstrated the same cytotoxicity of this extra sequence CAR-T cells, called CD19(86) CAR-T cells compared to parental CD19-CAR-T cells (20). The authors showed reduced cytokine secretion by these CAR-T cells (20). In addition, the authors performed phase I clinical trial and showed that CD19(86)-CAR-T cells had 54% complete remission of lymphoma with no CRS greater than 1 and no neurotoxicity observed in any of 25 patients treated (20). These data demonstrate that insertion of tag or other sequence extracellularly or intracellularly between ScFv and co-stimulatory and activation domain can change secretion of cytokines by CAR-T cells maintaining same cytotoxicity that is very valuable in clinic. The mechanism is not known but can include conformational, dimerization changes, changes of binding of adapter molecules mediating intracellular signaling and secretion of cytokines that needs to be studied in detail. It is clear that insertion of spacer between ScFv or elongating extracellular sequence and also insertion intracellular several amino-acids can change CAR-T cell mediated signaling that results in reduction of cytokine secretion. Further research into the effect of various epitope tags on ScFv conformation, CAR-T cytotoxicity, cytokine secretion and safety is of considerable interest.
The TF epitope tag has also proved useful for generating additional mock-CAR-T cells for experimental controls. We recently generated 3xTF mock CAR-T cells without an ScFv and transduced T cells with near 100% efficiency as detected by anti-TF antibody, although detection may have been improved by increased avidity through the incorporation of multiple epitopes. Nonspecific cytotoxicity of the 3x TF mock-CAR-T was similar to non-transduced T cells (data not shown) and may provide a better control than our prior mock-CAR-T in some instances. The data with half TF or 2xTF tag (30 extra amino-acids) support that insertion of different spacers between ScFv and signaling domains changes function of CAR-T cells.
In conclusion, this study shows application of an antibody epitope tag of human origin that may prove useful in CAR-T clinical studies. The tag allowed us to assess transduction efficiency and monitor CAR-T cells ex vivo. We observed high efficacy of CD19-TF and CD22-TF against CD19 and CD22-positive lymphoma cells. The cytotoxic effect was highly specific. These CAR-T cells had reduced cytokine secretion in vitro and even enhanced efficacy in the in vivo model. These findings suggest that TF epitope tagged CAR- may show superior efficacy and safety in clinical trial.
We would like to acknowledge Dr. Ed Lim for help with animal experiments and Xenogen Imaging. We would like also to acknowledge Promab scientists in Changsha, China for technical help with rabbit immunization. Each of the authors is employed by Promab Biotechnologies. Drs. Golubovskaya and Wu have filed for patent of CD19-TF-CAR-T cells.
CAR-T cells, CD19, CD22, Cytokine, Lymphoma, Immunotherapy.