
PolyADP-ribosylation is a post-translational modification which is involved in various physiological processes including maintenance of genome stability through DNA repair, regulation of transcription, and development. This process is also involved in pathological events such as cell death. Here, we review the effect of polyADP-ribosylation in signal transduction pathways in Drosophila melanogaster system. It is hoped that such an insight paves the way to develop therapeutics for human diseases.
PolyADP-ribosylation is a post-translational modification of proteins involved in various important physiological processes such as transcription, and in maintenance of genomic integrity and pathological conditions such as repair of DNA damage and cell death (1-2). PolyADP-ribosylation (or PARylation) is driven by a family of enzymes including poly(ADP-ribose) polymerases (PARPs) and ADP-ribosyl transferase 2 (ART2). These enzymes polymerize units of ADP-ribose moieties of NAD+ with alpha 1’’-2’ ribose-ribose linkages to form a long chain of ADP-ribose polymer [poly(ADP-ribose) or PAR] that occasionally show branching (Figure 1) (3-5). There are 17 PARP family members, which have sequence homology in their catalytic domain (6). PARP1, PARP2, PARP5A (tankyrase 1) and PARP6 (tankyrase 2) can form PAR, while other members either add only mono ADP-ribose residues (mono ADP-ribosylation or MARylation) to the acceptor protein or show no enzymatic activity (Figure 1). The enzymes that cause MARylation are also found in bacterial toxins (1). The nomenclature of family members of PARPs, their enzymatic activities and cellular localization have been recently reviewed (6).
 Figure 1
Figure 1
Synthesis and degradation of poly(ADP-ribose). PARPs, poly(ADP-ribose) polymerases; ART2, ADP-ribosyltransferase 2; PARG, poly(ADP-ribose) glycohydrolase; ARH3, ADP-ribosylhydrolase 3; TARG1: Terminal ADP-ribose protein glycohydrolase 1; Macro: MacroD1 and MacroD2; Na: nicotinic acid; Nam: nicotinamide; NaMN: nicotinic acid mononucleotide; NMN: nicotinamide mononucleotide; NaAD: nicotinic acid adenine dinucleotide; PAR: poly(ADP-ribose).
Understanding the function of polyADP-ribosylation in vivo was greatly advanced using Drosophila. Here, we discuss the importance of this model and review as how this understanding might be significant to the development of therapeutic strategies for human diseases. Drosophila melanogaster has Parp1, Parp5A (tankyrase 1) (HomoloGene-NCBI 1222 and 18405, respectively) and Parp16 genes (7).
Recent technological advances, especially mass spectroscopy, have allowed the identification of acceptor proteins that undergo polyADP-ribosylation and have unveiled the exact characterization of the amino acids that are modified by this process in these proteins. The original amino acids that were identified in the acceptor sites included glutamic acid, aspartic acid, lysine and arginine residues. However, using a filter-aided sample preparation approach combined with mass spectroscopy, Leidecker et al. recently reported that, in vivo, serine residues of histones are also targets for ADP-ribosylation (8). In addition to histones, PARP1 is also ADP-ribosylated at serine residues. Interestingly ADP-ribosylation of serine residues is observed only in the presence of histone polyADP-ribosylation factor 1 (HPF1/C4orf27) (9-10). Additional targets of serine ADP-ribosylation included high mobility group proteins and nucleophosmin (10). Consistent with involvement in stability of genome, it has also been shown that serine is the major target residue for ADP-ribosylation upon DNA damage (11).
Poly(ADP-ribose) (PAR) is hydrolyzed by PAR glycohydrolase (PARG) and ADP-ribosyl hydrolase 3 (ARH 3) (12-15) to form ADP-ribose residues (Figure 1). PolyADP-ribosylation was originally found in eukaryotic cells. However, Slade et al. demonstrated that filamentous fungi and a number of bacteria including Thermomonospora curvata have been shown to possess a divergent form of PARG that exhibits all the main characteristics of the human PARG. Based on the crystal structure of PARG in T. curvata, the catalytic domain of PARG was shown to be a distant member of the ubiquitous ADP-ribose-binding macrodomain family (16). Although mammalian PARG mainly hydrolyzes PAR chain, it can not cleave the bond that is formed between the proximal mono ADP-ribose and the amino acid residues of the acceptor proteins. In addition to ARH3 and TARG1/C6orf130, MacroD1 and MacroD2 can cleave the proximal mono ADP-ribose and the specified amino acid residues of the acceptor proteins (17-19) (Figure 1). Drosophila has Parg and Targ1/C6orf130 genes, however it is not reported to have Arh3 gene (HomoloGene-NCBI).
The genome structure of Parp1 of Drosophila was originally reported by Hanai et al. (20). Drosophila Parp1 (dParp1), which, corresponds respectively, to PARP1 and Parp1, in humans and mouse, is a single copy gene in haploid genome and has been mapped close to the centromeric heterochromatin regions of chromosome III 81F (20). The functional domains of dParp1 gene has six coding exons. Deletion mutants lacking all the coding exons of dParp1 were isolated and maintained in a heterozygous state; one allele being defective of dParp1 gene and the other allele being a balancer. Consistent with the Mendelian rule, crossing these heterozygous mutants should lead to a null mutant in one quarter of the offsprings. However, presumably because the homozygous deletion induced lethality, we failed to validate such a rate (21).
Tulin et al. reported that dParp1 gene spans more than 150 kb of a transposon-rich centromeric heterochromatin and produces several differentially spliced transcripts. Moreover, one non-coding exon was identified that is far upstream from the first coding exon in dParp1 (22). A novel isoform (Parp-e), which was expressed in embryo and ovary, was identified that possessed no enzymatic activity. An insertion mutation of P-element that is near the upstream promoter for Parp-e led to decreased Parp1 expression. This mutation also led to failure to form nucleoli and caused the heterochromatic sequence around dParp1 to become hypersensitive to micrococcal nuclease. These findings show that dParp1 is necessary for viability and plays a fundamental role in organizing chromatin structure including formation of nucleoli and heterochromatin (22). Although, a single PARP gene was originally described to be responsible for PARP activity in mammalian cells. Using Parp1 knockout mice, Amé et al. were able to show minor PARP activity. This led to their identification of a second gene (Parp2) in mice (23-24). Consistent with such results, the knockout of any of the family members of Parp in mammals has not led to the embryo lethality, while double knockout of mouse Parp1 and Parp2 was shown to cause lethality in embryos showing that, during development, these genes have mutually exclusive and important functions (25). In contrast to mice, in Drosophila Parp2 gene has not been identified.
For 6 hours after the eggs are laid, the level of mRNA of dParp1 is sustained at a high level while in subsequent two hours, its expression is gradually decreased. Then, for the following four hours, the expression increases again. The mRNA of dParp1 is not expressed in larvae while it is expressed in pupae and adult flies. At the early stage of embryo development, the abundance of the expression of Parp1 might be due to the presence of the maternal mRNA. Other than the pole cells, dParp1 is expressed uniformly in the embryos. This expression is gradually diminished until the stage 12, when expression of dParp1 mRNA gets restricted to the anal plates. At later stages of embryogenesis, dParp1 mRNA gets expressed in cells along the central nervous system (20).
The eye in Drosophila provides an excellent system for understanding cell differentiation, proliferation, and programmed cell death. By P element-mediated germ line transformation, Uchida M et al. overexpressed dParp1 in the embryos and in the primordia of the developing eye (26-27). The overexpression of dParp1 in the compound eyes led to the disruption of tissue polarity by improper rotation and chirality of the unit eyes. Each of the optical units that make up a compound eye (omatidia) had an abnormal shape, size and fusion and exhibited a mild disordered pattern (roughening). In addition, non-neuronal cells of midpupal retinas showed abnormal morphology and an asymmetrical arrangement. While the neuronal differentiation was normal, the overexpression of dParp1 led to the random orientation of photoreceptors in the eye disc of third instar larvae of the unit eyes. Moreover, in these eyes, the cytoskeletal F-actin was disorganized while some totally failed to show the normal accumulation of F-actin (26).
To test the effect of dParp1 in tissue polarity and cytoskeletal changes during embryogenesis and in adults, transgenic flies (hs-dParp1) were developed by adding a heat shock promoter to dParp1. Induction of overexpression of dParp1 by heat disrupted the tissue polarity and cytoskeletal organization and distribution of F actin in epidermis in stage 12-13 in developing embryos (27). Moreover, induction of this overexpression in adult flies led to similar changes in wing hair and in skin including the notum, abdomen and eyes (27).
Age-dependent accumulation of DNA damage leads to cellular senescence and reduces the overall age and lifespan of the organism. Since the classical Parp1 is thought to be involved in base excision repair and repair of double-strand DNA breaks, it follows that this gene is important to the lifespan (28-29). Shaposhnikov et al. reported that constitutive overexpression of dParp1 in the nervous system in Drosophila impacts the lifespan (30).
The idea that PARP1 genetically interacts with other genes was tested by genetic crosses between dParp1 transgenic flies and mutants or transgenic flies of other genes. RhoA is known to regulate the organization of cytoskeleton and tissue polarity during development. Overexpression of RhoA transgene in the eyes of Drosophila led to a rough eye phenotype and dramatically disrupted the architecture of the eye. However crossing of the RhoA transgenic flies with flies overexpressing dParp1, led to a reduction in the severity of the rough eye phenotype, suggesting of existence of some genetic interaction between dParp1 and RhoA (27).
In eukaryotic cells, ribosomes contain four types of RNA, designated as 5S, 5.8S, 18S, and 28S rRNAs. In these cells, nucleoli are the site for rRNA transcription, processing, and assembly. During this process, ribosomal precursor RNAs mature by virtue of binding ribosomal proteins to form complete ribosomal subunits that are transported to the cytoplasm. The 5.8S, 18S, and 28S rRNAs are transcribed as a single unit within the nucleolus by RNA polymerase I, resulting in the formation of a 45S ribosomal precursor that is then processed to the 18S rRNA of the 40S (small) ribosomal subunit and to the 5.8S and 28S rRNAs of the 60S (large) ribosomal subunit. 5S rRNA, which is also found in the 60S ribosomal subunit, is transcribed outside of the nucleolus, a process which is catalyzed by RNA polymerase III. Nucleoli are essential to cell growth (31). During each cell division, nucleolar organizing regions are assembled around the chromosomal regions by formation of nucleoli that contain the 5.8S, 18S, and 28S rRNA genes.
Boamah et al. reported that, when third instar larvae were cultured, for 12 hours in presence of a PARP inhibitor, 3-aminobenzamide, nucleoli were disintegrated and nucleoli-specific protein, fibrillarin showed aberrant localization. In addition, loss-of-function mutants of dParp1 showed a decreased level of ribosomes while increasing the 47S and 35S rRNA intermediates. Thus, dParp1 activity is required for targeting nucleolar proteins to the proximity of precursor rRNA. Thus, the activity of dParp1 activity is essential to nucleolar functions and ribosomal biogenesis (32).
It is suggested that PARP1 protein undergoes shuttling (32). In the first step of this process, most non-modified dParp1 protein binds to chromatin and accumulates in nucleoli. In the second stage, upon auto-modification with poly(ADP-ribose), dParp1 non-covalently interacts with a number of nuclear proteins forming protein-poly(ADP-ribose) complex that dissociates from chromatin into Cajal body (33).
“Leucine zipper” motif was proposed by Landschulz et al as repeating heptads of hydrophobic amino acid residues, usually leucine, which is associated with basic DNA binding region (designated as bZIP) (34). bZIP is found in transcription factors, such as C/EBP (CCAAT binding protein, Enhancer binding protein), c-Myc, N-Myc, L-Myc, v-jun, v-fos and yeast GCN4. Interestingly, dParp1 contains a leucine zipper with adjacent basic region, which fulfills the criteria of bZIP (34-35). While Jun and Fos contain five leucine repeats, C/EBP, CREB, yeast GCN4 and dParp1 contain four leucine repeats. However PARP1 in human, cow, mouse, chicken and Sarcophaga perigrina shows amino acid substitution in the corresponding regions (35-36). The auto-modification domain of PARP1 shows presence of “BRCT domains” that are involved in protein-protein interaction and are found in the DNA damage-responsive cell cycle checkpoint proteins (37). Kawamura et al. isolated an alternatively spliced form of dParp1 (designated as PARPII) that lacked the auto-modification domain (Figure 2) (38). This alternatively spliced form of dParp1 was isolated from stable transformant of Rat-1 cells which ectopically expressed PARPII under the regulation of MMTV-LTR promoter. As compared to cells transformed with the vector or full length dPARP1, after induction with dexamethasone, PARPII transformants showed reduced proliferation and exhibited loss of spindle shape (38). While GST-fusion dParp1 showed enzyme activity, GST-fusion PARPII had essentially no enzyme activity in presence of histone H1 or recombinant auto-modification domain (39).
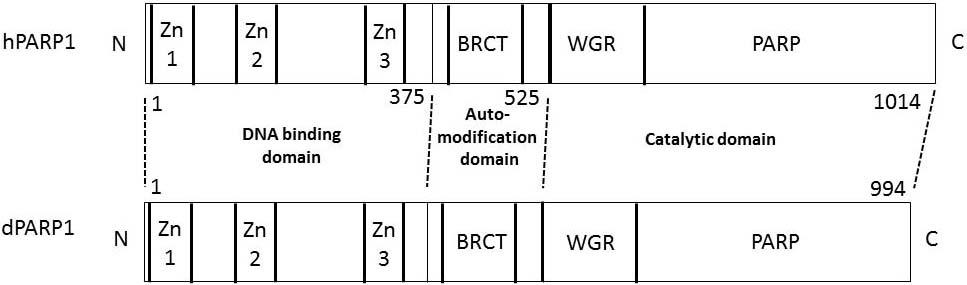 Figure 2
Figure 2
Domain structures of human PARP1 and Drosophila Parp1. The numbers refer to the amino acid number from the N-terminus at the junction of the domains in human PARP1. Zn1, Zn2, and Zn3 represent zinc finger domains 1, 2, and 3, respectively. BRCT, BRCA1 C terminus domain; WGR, tryptophan-glycine-arginine rich domain.
In Drosophila, the ribosomal proteins, L22 and L23a, which have N-terminal extensions, homologous to C-terminal portion of histone H1, might possess DNA-binding property, may be involved in organizing the ribosomes and can bind the auto-modification domain of dParp1 (40-41). Thus, in addition to being important to the organization of ribosomes, ribosomal proteins might also be involved in gene regulation and by interaction with PARP, ribosomal protein L22 and L23a might have other regulatory roles by impacting polyADP-ribosylation (42-44).
DNA transcription requires remodeling of chromatin structure and there is evidence that polyADP-ribosylation by PARP1 is involved in loosening of chromatin for active transcription of inducible genes. For example, in vitro, polyADP-ribosylation of chromatin (polynucleosomes) of calf thymus with PARP1, causes relaxation of chromatin whereas degradation of PAR by PARG leads to chromatin compaction (45-46). In similar in vitro experiments, d’Erme et al. showed that the chromatin from chicken erythrocytes relax after polyADP-ribosylation (47). Local loosening or puffing of polytene chromatin is required for induction of transcription of specific genes by external cues. Tulin and Spradling reported that puffing occurs at the defined chromosomal region in Drosophila larval salivary glands that are exposed to heat and that production of normal-sized puffs, requires both accumulation of hsp70 at these sites as well as the activity of dParp1 (48).
Pinnola et al. reported that the core histones, H3 and H4, are critical for binding of dParp1 to nucleosome. Histones H3 and H4 preferentially interact with the C-terminal portion of dParp1, while N-terminal domain of dParp1 negatively regulates these interactions. Importantly, the interaction of the H4 histone with the N-terminal tail, is required for induction of enzymatic activity of dParp1 in the absence of DNA (41).
The transcription of genes requires binding of the RNA polymerase complex to gene promoters and this binding requires local loosening of DNA. Post-translational modifications of the histones in the nucleosome might be essential to this process. For examples, in the Drosophila, the histone variant H2Av, which is the homolog of the mammalian histone H2A variants H2Az/H2Ax, co-localized with dParp1 in the hsp70 promoter region of third instar larvae. Subsequent phosphorylation of H2Av was required for stimulation of the enzymatic activity of dParp1, and the loosening of local chromatin for transcriptional activation of dParp1-target genes (49). JIL-1 kinase, which is an essential regulator of chromatin structure in Drosophila, phosphorylates H2Av within the nucleosome, leading to exposure of Val61 and Leu23 of the histone H4 in the nucleosome. This is critical for dParp1 binding to the nucleosome, polyADP-ribosylation of the surrounding chromatin proteins and chromatin loosening (50).
Hanai et al. by P element deletion, induced a loss-of-function mutant of Drosophila melanogaster that lacks the catalytic domain of Parg gene that encodes the major hydrolyzing enzyme of PAR (51). At normal developmental temperature of 25°C, the mutation caused lethality at the larval stages. Although, at 29°C, a quarter of the mutants reached adulthood, these animals had short lifespan and showed a progressive neurodegeneration that was exemplified by reduced locomotor activity. In brain of these mutants, there was an excess of PAR along with an extensive degeneration of neuronal cells that showed abnormal inclusion-like granules in the nuclei (Figure 3) (51). The regulation of PAR metabolism is important in the development and maintenance of homeostasis in the nervous system, since disruption of the murine Parg gene that leads to the failure to degrade PAR caused early embryonic lethality and enhanced sensitivity to genotoxic stress (52). Such a role has been suggested by identifying the biallelic mutations in ADPRHL2, encoding ARH3 in the “degenerative pediatric stress-induced epileptic ataxia syndrome” (53).
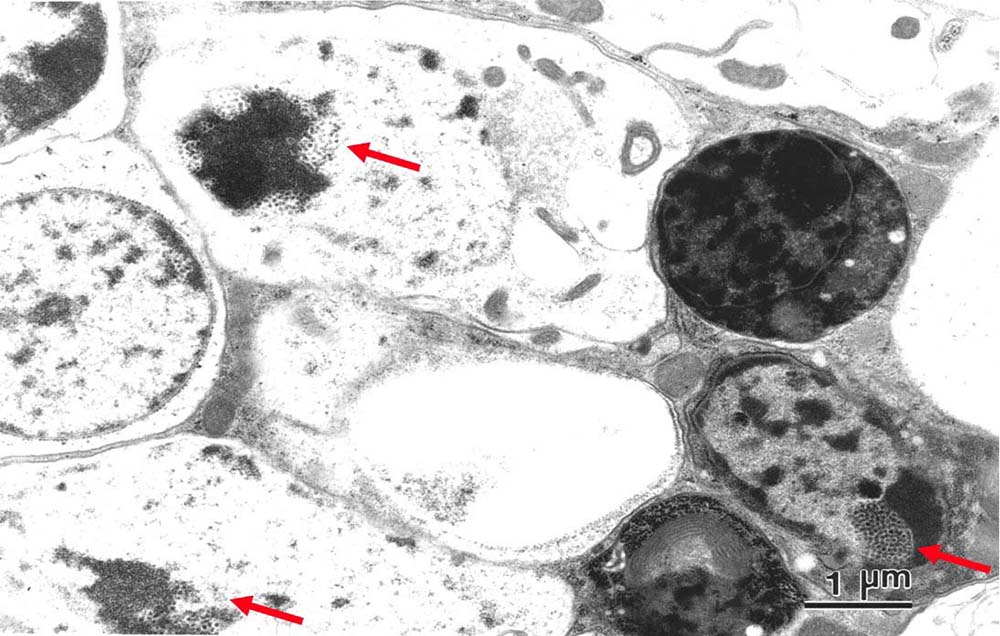 Figure 3
Figure 3
Electron microscopic view of horizontal section of an adult head of Drosophila melanogaster mutant Parg27.1./Y at 2 weeks after eclosion. The arrows show granular structures typical in the neural cells of the mutant. Reproduced with permission from (51) “Copyright (2003) National Academy of Sciences”.
Defect in the degradation of polyADP-ribosylated proteins could cause a disease that is manifested by accumulation of glutamyl ribose 5-phosphate in the lysosomes (Figure 1). This was shown in an 8-year-old boy who presented with a X-liked recessive inheritance of a form of lysosomal storage in the kidney and brain that, after 6 years, led to his demise from renal failure along with a progressive form of neurological degeneration (44-45). It was speculated that a defect in enzymes that catabolize polyADP-ribosylated proteins, such as ADP-ribose protein lyase, which remove the proximal ADP-ribosyl residue from histones or other acceptor proteins or alternatively the deficiency of terminal ADP-ribose protein glycohydrolase TARG1/C6orf130 can lead to a form of lysosome storage disease (Figure 1) (56-58).
Sharifi et al. recently reported a homozygous mutation of the C6orf130 gene in a family who showed a severe and progressive neurodegenerative disease associated with seizures without dysmorphic features (58). Due to occurrence of a premature stop codon, it was predicted that the mutation caused the formation of a truncated form of TARG1 protein that lacked the C-terminal half of the protein. They identified C6orf130 as a PARP-interacting protein that removes mono ADP-ribosylation on glutamate residues in PARP-modified proteins. Whole-exome sequencing, revealed a distinct homozygous sequence variant within exon 4 of the TARG1/C6orf130 gene that segregated with the neurodegenerative phenotype. Further analysis of the mutated C6orf130 suggested a transient C6orf130 lysyl-(ADP-ribose) intermediate. Depletion of the C6orf130 protein led to proliferation and DNA repair defects (58).
TARG1 protein was found to cleave mono ADP-ribosylated PARP 1 substrates (Figure 1). Since the bond between PARP1 and ADP-ribose in auto-modified PARP1 is glutamic acid, TARG1 has the enzymatic activity to cleave this bond. TARG1 differs from ARH3, which cleaves the bond between ADP-ribose and serine residue of the acceptor protein (15, 59). There are 58% identity and 74% similarity of amino acid sequences of the macro domains encoded by TARG1 genes in humans and flies. Almost full-length of the gene product is composed of single macro domain (HomoloGene-NCBI 17038).
PolyADP-ribosylation is important to the maintenance of genome, transcription, DNA repair and cell death. As shown in Figure 1, degradation of PAR is catalyzed by PARG and ARH3. However, while ARH3, TARG1, and Macro (D1, D2) can cleave the bond between mono ADP-ribose residue and the acceptor protein, PARG lacks such an activity.
Ghosh et al. identified a disease entity known as “Degenerative pediatric stress-induced epileptic ataxia syndrome” (53). Using linkage analysis and exome or genome sequencing in six families, they showed recessive inactivating mutations in ARH3 gene (ADPRHL2) in 5 of 6 exons. Although, after birth, patients were asymptomatic, they all showed developmental regression, and gradually developed progressive brain atrophy, spontaneous seizures, infections, and finally succumbed to a premature death. Three of 6 mutant proteins, expressed in E. coli, showed greatly reduced stability presumably due to conformational changes of the proteins (53).
In contrast to humans that have two genes (PARG and ADPRHL2 genes) that encode the enzymes that hydrolyze PAR, there is only a single gene (dParg) (NCBI HomoloGene: 9863 and 50532) in the Drosophila that likely carries all the functions of PARG and ARH3. There are findings that suggest that human ADPRLH2 is a functional paralog of Drosophila Parg. For example, the siRNA knockdown of dParg decreased survival and increased susceptibility to exogenous stresses such as hydrogen peroxide. The enzymatic activity of PARP1 is directly inhibited by minocycline as well as other tetracycline derivatives (60) that exert neuroprotective and anti-inflammatory actions. In flies with ubiquitous and neuronal knockdown of dParg, pretreatment with minocycline, increased survival of the flies in presence of hydrogen peroxide (53). Forced expression of human ADPRHL2 gene in two different transgenic lines also promoted survival and prevented defect of locomotor activity in Drosophila with dParg knockout (51,53). Thus, inhibitors of PARP1 might be therapeutically useful to prevent neurodegeneration.
In a large international survey, a truncating mutation in alcohol dehydrogenase gene (ADH) class1 (ADH1C (G78Stop)) showed significant association with Parkinson’s disease (61). Moreover, knockout of Adh class1 (Adh1) and class 1 and class 4 genes (Adh1/4) in mice led to phenotypes that were similar to the Parkinson’s disease. These animals showed reduced weight, locomotion, smell, and reduced monoamine levels (62). The peroxidation of polyunsaturated fatty acids through lipid peroxidation, can lead to the formation of toxic levels of aldehydes that can be reduced by ADH, especially ADH1 (63-64). Thus, it follows that knockout of Adh1 and Adh1/4 genes should lead to the accumulation of toxic levels of aldehydes. Among these aldehydes, acetaldehyde can react with dopamine, forming salsolinol (65), a neurotoxin with an effect similar to the effects of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyrimidine (MPTP) that is known to cause parkinsonism (66). Once transferred to the brain, these compounds can cause degeneration of dopamine neurons in the substantia nigra through inhibition of the mitochondrial respiratory chain complex I (67).
One of the typical pathological findings in patients with Parkinson’s disease is the accumulation of fibrils in the brain which are formed from α-synuclein. Recently, Kam et al. reported that in mouse primary cortical neurons, neurotoxicity induced by α-synuclein was dependent on polyADP-ribosylation (68). We recently reported that in Parg mutant Drosophila (51), which showed neurodegeneration, ADH was a major acceptor protein for polyADP-ribosylation (69). Thus, we argue that if polyADP-ribosylation of ADH1 inhibits its enzymatic activity, then a PARP inhibitor should theoretically increase ADH activity. Consistent with this, pharmacological inhibition of PARP with olaparib (70), led to a significant increase of ADH activity in a human hepatoma cell line (69). In line with these findings, dParp1 mutation rescued parkin and pink1 mutant phenotypes of Drosophila (71-72).
PolyADP-ribosylation and its involvement in the DNA repair and tissue homeostasis offers unique opportunities for the development of strategies for the treatment of cancers and neurodegenerative diseases. In this respect, it has been shown that cell death can be caused by inhibition of PARP in breast and ovarian cancer cells that carry mutation of BRCA1/2, which is important for repair of homologous recombination (73-75). Moreover, the loss-of-function mutant of Drosophila, e.g. Parg and C6orf130, has been suggested to be a useful screening tool in therapeutics, and in some hereditary neurodegenerative diseases (58).
This work was supported partly by Grants-in-Aid for Scientific Research (23590350) from the Japan Society for the Promotion of Science, Japan. We would like to thank Dr. Yumi Koyama, Dr. Tomonori Kawamura and the late Dr. Masahiro Uchida of Institute of Basic Medical Sciences, University of Tsukuba, Japan, Mr. Yasuhito Kuroda, Mr. Hiroto Nodono, Ms. Akiko Hamada and Mr. Yutaka Nakatani of Nagahama Institute of Bio-Science and Technology, Japan, and Dr. Palmiro Poltronieri, Institute of Sciences of Food Productions, National Research Council of Italy (CNR-ISPA), Italy, for supporting this work.