
Vγ9Vδ2 T cells are immune effector cells very well-suited for immunotherapy, but clinical results have been disappointing in multiple myeloma (MM) and other cancers. We have shown that Vγ9Vδ2 T cells are victimized prematurely by the immune suppressive tumor microenvironment (TME) established by myeloma and neighbouring cells in the bone marrow (BM) of MM patients. One major mechanism is the highly redundant expression of multiple immunecheckpoints/immune checkpoint-ligands (ICP/ICP-L) in the TME impairing antimyeloma Vγ9Vδ2 T-cell immune responses. Another major immune suppressive mechanism is the metabolic reset driven by myeloma cells in the TME to satisfy their energetic needs to the detriment of effector cells. Recently, it has become clear that the ICP/ICP-L circuitry and metabolic checkpoints (MCP) jointly operate in the TME of cancer patients to promote tumor cell growth and suppress antitumor immune responses. In this review, we discuss the possible interactions between ICP/ICP-L and MCP in the TME of MM patients that may compromise the immune competence of BM Vγ9Vδ2 T cells, envisaging novel combination therapies to improve the outcome of immune-based interventions.
Vγ9Vδ2 T cells are attractive candidates for ex-vivo and in vivo adoptive immunotherapy based on their capacity to bridge innate and adaptive immunity and to exert a multifaceted array of direct and indirect antitumor immune responses (1). Several in vitro data indicate that myeloma cells are privileged targets of Vγ9Vδ2 T cells, but clinical trials have fallen short of clinical expectations (2-4). We have shown that bone marrow (BM) Vγ9Vδ2 T cells are very susceptible to the immune suppressive context created by myeloma cells in the tumor microenvironment (TME) and very refractory to regain their immune competence status (5).
Vγ9Vδ2 T-cell dysfunction largely anticipates that of other immune effector cells, like CD8+ and CD4+ cells, indicating that, the initial myeloma cell expansion strategy, from the immunological standpoint, is aimed at neutralizing immune cells with the highest capacity to jeopardize myeloma cell growth and survival (5). The PD-1/PD-L1 axis is deeply involved in the neutralization of BM Vγ9Vδ2 T cells in MM (6,7), but single PD-1 blockade is insufficient to fully rescue in vitro the immune functions of Vγ9Vδ2 T cells (5). These data recapitulate the discouraging results of clinical trials in MM and other cancers (8, 9). Preliminary results from our laboratory indicate that the TME in MM patients is characterized by the expression of multiple immune checkpoints/immune checkpoint-ligands (ICP/ICP-L) in myeloma cells, immune effector cells, immune suppressor cells, bone marrow stromal cells (BMSC), and endothelial cells. This highly redundant ICP/ICP-L expression creates a very resilient immune suppressive network almost impossible to overcome by single ICP/ICP-L blockade.
The ICP/ICP-L circuitry is not the only molecular machinery involved in the establishment of the immune suppressive TME in MM and other cancers. Tumor cells must reprogram their metabolic pathways to support their expansion strategy against normal cells and immune cells in a microenvironment characterized by suboptimal vascularization and limited nutrients supply. To this end, tumor cells work for an increased rate of glycolysis and over-utilization of amino acids (i.e., glutamine, arginine, and tryptophan) resulting in the increased production of toxic waste in the TME. The combination of key nutrients deprivation and accumulation of immune suppressive metabolites (i.e., lactate, kynurenine, and adenosine) compromises the capacity of immune effector cells to mount effective antitumor immune responses as summarized in Figure 1 (10,11).
 Figure 1
Figure 1
Metabolic alterations in the TME: consequences on tumor cells and immune cells. The energy supply needed by tumor cells to secure their survival and expansion in the TME is met by the enhanced utilization of nutrients locally available (i.e., glucose and amino acids). Far from being ecologically sustainable, the metabolic changes operated by tumor cells in the TME lead to the release of toxic waste that are responsible for the induction of a progressively hypoxic, acidic, and poor in nutrients TME. The immune cells respond in a different way to these challenges: toxic metabolites (i.e., lactate, kinurenines, Ado, etc) promote the activation of suppressor cells like Tregs, MDSC, M2 macrophages, whereas they restrain the activation of immune effector cells (Teffs). The metabolic reset operated by tumor cells (i.e., aerobic glycolysis, up-regulation of lipid/amino acid metabolism, etc) allows tumor cells to forestall the limited energetic resources locally available, whereas effector T cells are unable to reset their metabolism and fail the competition to secure the nutrients needed to support their activation. Immune suppressor cells also contribute to brake the immune reactivity of Teffs, that become functionally exhausted. The hypoxic and acidic features of the TME also contribute to the genetic and epigenetic instability of tumor cells promoting clonal evolution and immune escape. Many of the metabolic alterations summarized in this Figure have also been reported in the BM of MM patients.
The metabolic alterations occurring in the TME act as metabolic checkpoints (MCP) limiting not only the physiological immune surveillance exerted by innate and adaptive immunity, but also the efficacy of therapeutic immune interventions including ICP/ICP-L blockade. The rescue of immune effector cells by anti-PD-1 monoclonal antibodies (mAb) can be thwarted if the local TME is unfit to support the energetic needs of these cells once freed from the brake imposed by the PD-1/PD-1L network. So far, the most common strategy exploited to overcome inadequate ICP/ICP-L blockade has been the combination of multiple ICP/ICP-L inhibitors, but this approach is limited by the prohibitive costs and increased side effects. Our data in MM patients provide further evidence that the ICP/ICP-L circuitry is highly redundant and almost impossible to be resolved by single ICP/ICP-L blockade.
Mutations that activate oncogenic signaling pathways are progressively used as combination targets in onco-immunology to improve the efficacy of ICP/ICP-L blockade (12). An alternative approach is to target the interplay between ICP/ICP-L and MCP. Considerable efforts are underway to elucidate the mechanisms that MCP and ICP/ICP-L jointly operate in the TME to promote tumor cell growth and suppress antitumor immune responses (13).
This review will discuss how MCP can compromise the antitumor functions of immune effector cells, including Vγ9Vδ2 T cells, and whether targeting the affected pathways in combination with ICP/ICP-L blockade can be a powerful and cost-effective approach to enhance antitumor immune responses. We believe that the BM of MM patients is a very informative training-ground to address this issue and formulate novel hypotheses of therapeutic interventions based on concurrent MCP and ICP/ICP-L blockade.
The BM of MM patients is not exempted from the metabolic alterations occurring in the TME of other hematological and solid tumors (14,15). This metabolic reprogramming is finalized to meet the energetic needs of myeloma cells to the detriment of bystander cells with special regard to immune effector cells (14,16). Enhanced glycolysis (17) has been reported in myeloma cells to provide either fuel and/or building blocks for their survival and proliferation. Several mechanisms are involved in sustaining the enhanced glycolysis of myeloma cells. Hexokinase II (HKII), one of the pacemaker-enzyme of glycolysis, is constitutively over-expressed in myeloma cells, and HKII inhibition with 3-bromopyruvate (3BrPA) strongly suppresses ATP production leading to myeloma cell death (17). Pyruvate kinase M2 (PKM2) is another key player involved in the regulation of aerobic glycolysis to sustain tumor cell proliferation and survival. Alternative splicing of PKM isoforms by NIMA (never in mitosis gene A)-related kinase 2 (NEK2) promotes aerobic glycolysis in myeloma cells by increasing the PKM2/PKM1 ratio (18). Aerobic glycolysis is particularly accelerated in myeloma cells from patients with high-risk MM that are characterized by the aberrant expression of LDHA, PDK1 and GLUT1 (19).
Glutaminolysis is also increased in myeloma cells (14,20,21). Myeloma cells display high expression of glutamine transporters and are highly sensitive to glutamine depletion suggesting a glutamine-addicted status (20,22).
The toxic waste (e.g. lactate) generated by the increased utilization of glucose by myeloma cells and the accumulation of ATP breakdown products like adenosine (Ado) create a slightly acidic and hypoxic TME which, in combination with key nutrients depletion, promotes tumor cell survival and suppress antitumor immune responses (23-25).
Physiologically, the BM is already more hypoxic than the peripheral blood (26). This constitutive tendency to hypoxia is exacerbated in hematological malignancies arising in the BM like MM (27,28). Most changes induced by hypoxia are mediated by the transcription factor hypoxia-inducible factor-1 (HIF-1), consisting of HIF-1α and HIF-1β. The former is a cytosolic subunit degraded under normoxia, but which translocates into the nucleus under hypoxia; the latter is oxygen-insensitive protein constitutively present in the nucleus where it acts as HIF-1α co-activator. The dimer HIF-1α/HIF-1β, together with other isoforms such as HIF-2, is responsible for inducing several transcriptional programs in the hypoxic TME (27,28).
Interest in this area has been raised by the recognition that angiogenesis and disease progression are associated in MM and that hypoxia is a critical regulator of angiogenesis (29). Subsequent studies have confirmed that the BM of MM mouse models and MM patients is hypoxic compared to healthy controls (30,27) and that hypoxia plays a crucial role in triggering the metabolic alteration that are needed to sustain the growth of myeloma cells and promote their resistance to chemotherapy (27, 31-33).
Maiso and colleagues (34) have reported that the disease relapse in MM is initiated by residual myeloma cells which have become resistant to chemotherapy and are strategically located in the hypoxic niches of the TME. Drug resistance is gained via HIF-1α activation and up-regulation of the glycolytic enzymes HKII and lactate dehydrogenase A (LDHA). Development of melphalan resistance has also been associated with increased aerobic glycolysis and elevated oxidative stress response mediated by VEGF/IL8-signaling. In addition, up-regulated aldo-keto reductase levels of the AKR1C family involved in prostaglandin synthesis contribute to the resistant phenotype (35).
As expected, hypoxia has become a therapeutic target in MM and several approaches have been investigated in MM mouse models and human MM (28,30,36). Maiso et al have shown that the genetic knock-down of HIF-1α and/or LDHA restores the sensitivity of myeloma cells to bortezomib and melphalan (34). Glutaminolysis can also be involved in drug resistance since glutaminase 1, the pacemaker enzyme of glutaminolysis, is increased in bortezomib-resistant myeloma cell lines compared to sensitive cells (21). The anaplerotic flux driven by glutaminolysis fuels the tricarboxyclic acid (TCA) cycle and the mitochondrial oxidative phosphorylation (OXPHOS) to produce huge amounts of ATP. The selective glutaminase 1 inhibitor CB-839 reduces the availability of ATP and restores bortezomib sensitivity (37).
Very comprehensive reviews about the metabolic pathways that are significantly altered in myeloma cells vs normal B cells have recently been reported (38, 15). The hexosamine pathway, the amino acid interconversion reactions and degradation processes, the folate and the methionine cycle, the cholesterol uptake and metabolism, and the fatty acid synthase (FAS) are up-regulated especially in myeloma cells from MM patients with advanced disease.
Interestingly, the metabolic alterations occurring in the TME of MM patients are so robust to be mirrored in the peripheral blood. Steiner et al. have shown that the plasma levels of several metabolites derived from the amino acid, lipid, and classical catabolic pathways are significantly different in healthy volunteers compared to individuals with monoclonal gammopathy of undetermined significance (MGUS) and MM patients. IDO-driven immune regulation and glutaminolysis have emerged as the most promising therapeutic targets from this metabolomic analysis (16). Similar results have been reported by Du H et al. who have performed non-targeted metabolomic analyses on serum samples derived from responder and non- responder MM patients in comparison with healthy controls. The aim of the study was to identify non-invasive biomarkers for diagnosis and clinical monitoring of MM patients. They have identified 10 metabolites, mainly representative of the arginine/proline and glycerophospholipid metabolism, which were significantly different among groups and correlated with clinical outcome (39).
The metabolic changes occurring in the TME of MM patients are also responsible for the activation of pathway allowing myeloma cells to evade immune cell recognition and killing. Examples are the up-regulation of CD38, CD46, SLAMF7 (CS1), and ICP/ICP-L on myeloma cells and bystander cells (38). PKM2 has been reported to induce HIF-1α transactivation and recruitment of the hypoxia response elements (HRE) of HIF-1α target genes including PD-L1 on macrophages, dendritic cells (DCs), T cells, and tumor cells (40). Lactate and Ado-derivatives like cAMP generated by tumor cells in hypoxic TME can induce arginase (Arg-1) and indoleamine-2,3-dyoxigenase (IDO) activation in antigen presenting cells (APC). Arg-1 induced depletion of arginine, an essential amino acid to sustain T-cell proliferation, and arginine catabolism into urea and ornithine hampers the expansion of antitumor effector T cells. In parallel, kynurenine produced by IDO, that is up-regulated in the TME of MM patients, reinforces the inhibition of effector T cells and promotes the differentiation of regulatory T cells (Treg) (25). APC, myeloma cells and BMSC are the major IDO sources in the MM TME (41, 42) making this pathway an attractive metabolic target to increase the efficacy of antitumor immunotherapy (43).
The mevalonate (Mev) pathway has gained a great interest in the field when it was found that zoledronic acid (ZA), the most potent aminobisphophonate (NBP) commonly used to treat bone disease in MM and other solid cancers, can induce Vγ9Vδ2 T-cell activation. The mechanism consists in the inhibition of farnesyl-pyrophosphate synthase (FPPS) and accumulation of isopentenylpyrophosphate (IPP). IPP is structurally related to the phosphoantigens (pAgs) generated by mammalian and non-mammalian cells that Vγ9Vδ2 T cells recognize as part of their natural duty to react against pathogens, stressed cells, and tumor cells. Mev pathway up-regulation in myeloma cells is one reason why Vγ9Vδ2 T cells have a natural predisposition to recognize and kill myeloma cells in vitro (44, 45). As a result of the increased Mev pathway dysregulation, myeloma cells endogenously synthesize and release high amounts of IPP.
Mev is synthesised intracellularly from the 3-hydroxy-3-methylglutaryl coenzyme A (HMGCoA) in a process catalysed by the rate-limiting enzyme HMG-CoA reductase (HMGR). The Mev pathway converts mevalonate into sterols (i.e, cholesterol) and nonsterol isoprenoids [i.e, FPP and geranylgeranylpyrophosphate (GGPP)]. Isoprenoids are critical for the isoprenylation of monomeric G-proteins such as Ras, Rho and Rac that are essential for cell growth and migration. FPP is specifically generated by FPPS, the enzyme targeted by ZA downstream to HMGR. The metabolic consequences of ZA-induced FPPS inhibition are both a reduced content of intracellular FFP-derived isoprenoids (leading to the impaired activity of prosurvival Ras-dependent pathways) (46), and intracellular IPP accumulation (47).
Intracellular IPP accumulation may have different fates depending on the cell capacity to handle the overload. In cells like osteoclasts and selected tumor cells, IPP is combined with AMP by aminoacyl-tRNA-synthetase and transformed into the pro-apoptotic ATP analog 1-adenosin-5-yl3-(3-methylbut-3-enyl) triphosphoric acid diester (ApppI) (48). ApppI accumulation induces apoptosis which is the mechanism exploited to treat bone disease in MM and cancer patients with bone metastases. On the contrary, cells like DC and BMSC are metabolically equipped to escape ApppI accumulation and apoptosis. We have shown that ATP-binding cassette transporter A1 (ABCA1) is involved with BTN3A1 and apoAI in the extrusion of intracellular IPP in DC, BMSC and other cells when IPP concentrations exceed a critical threshold (31,32). In these cells, the ABCA1/BTN3A1/apo-AI complex acts as safety valve to prevent ApppI formation and protect cell viability. The consequence of extracellular IPP can be different according to the releasing cells (i.e., DC vs BMSC) and the local microenvironment (i.e., TME vs secondary lymphoid organs) with the consequence to drive different functional outcomes in Vγ9Vδ2 T cells (i.e., activation vs functional exhaustion) (49,50).
The Mev pathway can also be targeted by statins that are potent HMGR inhibitors. By acting upstream to FPPS, statins suppress the entire pathway without inducing intracellular IPP accumulation. Both statins and ZA may have direct antitumor effects by inducing apoptosis and inhibition of myeloma cell proliferation (51-54). Interestingly, cytotoxicity of ZA against cancer cells is potentiated by hypoxia, via the loss of HMGR activity, suggesting that these drugs can outperform in cancer patients under hypoxic conditions (55).
Statins and ZA have also been shown to decrease P-glycoprotein-dependent multidrug resistance by reducing the Ras/ERK-dependent activation of HIF-1α (56,46). Mev pathway inhibition with simvastatin and lenalidomide has demonstrated a synergistic antimyeloma activity suggesting that this combination is worth investigating in the treatment of relapsed/ refractory MM (57). Unlike ZA, statins cannot activate Vγ9Vδ2 T cells because the Mev pathway inhibition is up-stream to FPPS and there is no IPP accumulation. Thus, ZA only can be used to intentionally activate antitumor immunity mediated by Vγ9Vδ2 T cells (58, 47) and NK cells (59,60). However, Vγ9Vδ2 T-cell based immune interventions in MM have not met clinical expectations. Our interpretation is that the protumoral partnership operated by ICP/ICL and MCP has been underestimated and the strategies fielded to rescue Vγ9Vδ2 T-cell functions inadequate.
Very few data are available about the impact of TME metabolic alterations on Vγ9Vδ2 T cells. Figure 2 illustrates the expected interplay between metabolic alterations occurring in myeloma cells and immune cells in the TME of MM patients. It is very unlikely that Vγ9Vδ2 T cells can escape the metabolic constraints elaborated by myeloma cells and bystander cells in the TME of MM patients. As reported above, Vγ9Vδ2 T cells are more susceptible to immune suppression than other immune effector cells as shown by PD-1 expression and anergy to pAg stimulation already detectable in MGUS individuals, when the BM myeloma cell infiltration is minimal (< 10% by definition) and the disease totally asymptomatic.
 Figure 2
Figure 2
Metabolic pathways involved in the immune suppressive TME of MM patients. Anaerobic and aerobic glycolysis, fatty acid β-oxidation, glutaminolysis, and oxidative phosphorylation are metabolic pathways supplying energy and building blocks to myeloma cells (MM cells) The mevalonate pathway (Mev) provides building blocks (cholesterol, isoprenoids) and phosphoantigens (i.e. IPP) that can activate or inhibit Vγ9Vδ2 T cells depending on local concentrations. ATP-degradation via CD39 and CD73, arginine and tryptophan catabolism via Arginase-1 (Arg-1) and indoleamine 2,3-dioxygenase (IDO) are catabolic pathways operative in myeloma cells and neighboring cells (myeloid-derived suppressor cells, MDSC; bone marrow stromal cells, BMSC) that contribute to the generation of the immunosuppressive tumor microenvironment (TME). Hypoxia and the transcription factor hypoxia inducible factor-1α (HIF-1α) promote anaerobic glycolysis and up-regulate the immune-checkpoint (ICP) PD-L1 and the ATP binding cassette transporter ABCA1. ABCA1 in cooperation with apolipoprotein A-I (apoA-I) and butyrophilin-3A1 (BTN3A1) extrudes supra-physiological IPP amounts from myeloma cells and BMCS leading to Vγ9Vδ2 T-cell exhaustion.
Like conventional T cells, Vγ9Vδ2 T cells need to reset their metabolism when they are engaged by specific challenges and must mount an effective immune response. Vγ9Vδ2 T cells can also be divided into subsets according to their phenotype, proliferative capacity and effector functions. Naïve and memory Vγ9Vδ2 T cells (both CD27+) display high proliferative capacity, but low effector functions, whereas effector and late effector Vγ9Vδ2 T cells (both CD27-) display the opposite pattern (61). Naïve T cells oxidize glucose-derived pyruvate, along with lipids and amino acids, to efficiently produce ATP/energy required for immune surveillance. Upon activation, fatty acid oxidation (FAO) is down-regulated, and glycolysis and glutaminolysis up-regulated to quickly produce enough energy to sustain the rapid cell growth and proliferation requirements. At the end of the immune response, cells committed to become memory T cells revert to FAO which is more efficient to generate energy under less stressful conditions (62).
This metabolic reset is challenged in the TME which is intrinsically hostile to the immune system and metabolically committed to promote tumor cell survival, restrain antitumor immune responses, and concurrently boost protumoral immune responses as illustrated in Figure 1 and Figure 2.
No data are available about the metabolic reset operated by Vγ9Vδ2 T cells challenged by tumor cells in the TME, but some interesting speculations can be inferred from experimental models and the behavior of other innate effector cells. Laird et al have investigated glucose metabolism in a Listeria mouse model of infection and shown that γδ T cells express higher surface levels of glucose transporters than αβ T cells. γδ T cells exhibit effector functions over a broader range of glucose concentrations after activation, suggesting a greater dependency on glucose uptake and metabolism than activated αβ cells (63). This metabolic behavior is reminiscent of what NK cells operate to boost aerobic glycolysis and sustain IL-15-driven differentiation and activation (64). The greater γδ T-cell dependency on glycolysis may explain why the dysfunction of Vγ9Vδ2 T cells is already detectable in MGUS and anticipates the immune dysfunctions of CD4+ and CD8+ cells.
Another major MCP constraint limiting the antitumor activity of CD8+ T cells and NK cells (65, 66) is amino acid depletion which reinforces the immune suppressive effects of glucose deprivation in the TME (67-69). Glutamine deprivation suppresses T-cell proliferation and cytokine production by preventing Th1-cell differentiation, while fostering Treg development (70). We are currently investigating whether the glutamine deprivation reported in the TME of MM patients (22) can contribute to Vγ9Vδ2 T-cell dysfunction and whether strategies preventing glutamine deprivation can help to rescue their immune competence.
Alterations in arginine metabolism also have a critical role in modulating antitumor immune responses. L-Arginine is necessary to promote T-cell cycle progression and switch from glycolysis to oxidative phosphorylation (OXPHOS), and to sustain the generation of memory T cells (71). L-Arginine is a critical energy resource for effector T cells in a glucose-deprived TME, but the TME operates to minimize its concentrations and blunt T-cell activation. Tumor cells produce metabolites (i.e, cAMP, lactate or HIF-1α-induced molecules) and cytokines (i.e, IL-4, IL-6, IL-13, M-CSF or GM-CSF) that up-regulate arginase 1 (Arg1) expression in tumor-associated macrophages and MDSC leading to L-arginine degradation and suppression of T-cell effector functions (72). This immune suppressive mechanism is operated by myeloma cells and MDSC in the TME of MM patients (73). Sacchi A. et al. have argued that Arg1 up-regulation and L-arginine deprivation can be involved in the immunosuppression of Vγ9Vδ2 T cells mediated by MDSC (74). Accordingly, Arg1 inhibition restores the ability of Vγ9Vδ2 T cells to produce IFN-γ production and kill Daudi and Jurkat cells overcoming MDSC-induced immunosuppression (74).
Another major MCP is the increased tryptophan catabolism mediated by IDO and leading to increased kynurenine levels in the TME. On the one hand, tryptophan deprivation impairs T-cell proliferation and cytotoxic T-cell responses (75); on the other, the overproduction of kynurenines in the TME induces Treg differentiation and suppresses T-cell effector functions by downregulating the T-cell receptor CD3 ζ chain (76,77). Like other MCP, tryptophan catabolism P operates in the TME to tip the balance in favor of Treg to the detriment of effector T cells. Based on these data, it is not surprising that Treg are steadily rooted in the BM of MM patients irrespectively of the disease status and never loose their capacity to blunt antimyeloma immune responses, including those mediated by Vγ9Vδ2 T cells (78). Interestingly, Kunzmann et al. have reported that Treg can specifically suppress Vγ9Vδ2 T cells in MM patients (79). The uncontrolled production of kynurenine by IDO, largely overexpressed in the MM TME (41,42), can also contribute to Vγ9Vδ2 T-cell immune suppression. Data by Fechter K et al support this hypothesis since they have shown kynurenines produced by BM mesenchimal stem cells can affect Vγ9Vδ2 T-cell activation (80).
Hypoxia is another major hurdle to antimyeloma immune responses mediated by Vγ9Vδ2 T cells. HIF-1α can promote the generation and maintenance of Treg cells (81) and induce the expression of PD-L1 in tumor cells and MDSC (82-85). We have reported that MDSC are PD-L1+ in the TME of MM patients and these cells do not fade away even when almost all myeloma cells have been eliminated by autologous stem cell transplantation (5). Very few data are available about the effect of hypoxia on Vγ9Vδ2 T-cell functions. Siegers GM and colleagues (86) have found an increased infiltration of Vγ9Vδ2 T cells in the hypoxic areas of breast cancer patients, but MIC-A shedding protects breast cancer cells from Vγ9Vδ2 T-cell recognition and killing. More recently, it has been shown in vivo that MDSC can modulate the cytotoxicity of γδ T cells via tumor-derived exosomes engaged by the miR-21/PTEN/PD-L1 axis (87). We have initiated to evaluate the effect of hypoxia on pAg-reactivity of normal Vγ9Vδ2 T cells and found that it is almost completely abrogated. We are currently investigating whether this is a direct consequent of HIF-1α activity or a consequence of HIF-1α-driven metabolic reprogramming.
Another major MCP which can restrain Vγ9Vδ2 T-cell functions is Ado, whose concentrations are elevated in the TME of MM patients due to the coordinated expression of adenosinergic ecto-nucleotidases (CD39/CD73/CD38/CD203a) strategically located in the hypoxic BM niches (88, 89). The immune suppressive effect of Ado on conventional effector T cells is well known (90), whereas no data are available onVγ9Vδ2 T cells. Data in a mouse model of experimental autoimmune uveitis indicate that γδ T cells (not Vγ9Vδ2 T cells because mice lack this subset) have an increased expression of high-affinity adenosine receptors (A2ARs) and that A2AR ligation inhibits αβ T cell activation, but enhances γδ T cell activation (91). The high A2ARs expression enables γδ T cells to sequester Ado, prevents Treg expansion, and unleashes αβ T-cell activation (92). Whether similar interactions between Ado, Treg, conventional αβ T cells and Vγ9Vδ2 T cells also occur in humans is unknown. We have previously reported that ZA induces an advantageous cross-talk in MM between Vγ9Vδ2 T cells, αβ CD8+ T cells, regulatory T cells, and DC, but these data were obtained using PB cells and not cells isolated from the TME (47).
An additional layer of metabolic complexity that Vγ9Vδ2 T cells have to cope with in the TME is due to their unique ability to recognize pAgs generated in the Mev pathway of tumor cells and bystander cells. We have shown that BMSC in the TME of MGUS and MM patients release huge amounts of IPP which can be regarded as a MCP highly specific for Vγ9Vδ2 T cells. We have shown that IPP release in the TME by BMSC is an early event already detectable in MGUS individuals. This long-lasting exposure to IPP can be responsible for the early senescence experienced by Vγ9Vδ2 T cells in the TME of MGUS and MM (6). These data are in keeping with the serum metabolomic profiles of MGUS (see above) which already different compared with healthy controls indicating that some metabolic alterations occur very early in the biology of MGUS/MM evolution similar to the dysfunction affecting Vγ9Vδ2 T cells.
Immunotherapy in cancer has finally burst on the clinical scene after being questioned for many years. Most of the breakthrough has been triggered by monoclonal antibodies (mAbs) blocking ICP/ICP-L interactions, but several issues remain to be solved to fully exploit this long-sought therapeutic opportunity. It is not known which are the most effective immune cells to be rescued from immune suppression and to be redirected against tumor cells (i.e., CD8+ cells, NK cells, NKT cells, Vγ9Vδ2 T cells, etc). Likewise, it is not known which is the best combination therapy since single PD-1/PD-L1 blockade is inadequate in many cancers, including MM. The metabolic alterations occurring in the TME of MM patients are therapeutic targets very promising because of the interplay with ICP/ICP-L and their potential druggability using small (and often inexpensive) molecules which are very well suited to target intracellular pathways (93-95).
Targeting MCP in combination with ICP/ICP-L blockade can enhance antimyeloma Vγ9Vδ2 T-cell functions and improve the efficacy of immune-based interventions in MM and other cancers. Speculative combination strategies are discussed below and graphically summarized in Figure 3.
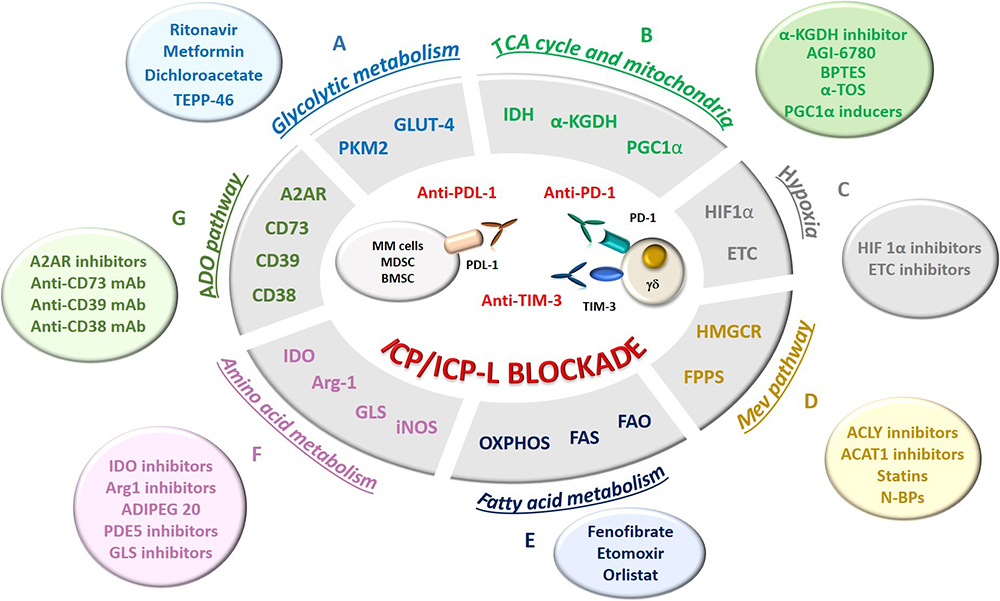 Figure 3
Figure 3
Possible strategies to rescue Vγ9Vδ2 T cells by targeting MCP in combination with ICP/ICP-L blockade in MM. MCP are shown in the grey ellipse; the relative activatory/inhibitory molecules are shown in the colored circles. A. Glycolytic and intermediate metabolism (light blue): myeloma cells exhibit high expression of glucose transporter 4 (GLUT4) for basal glucose consumption, whose uptake can be override by HIV protease inhibitor ritonavir. The combination of ritonavir and metformin can improve antimyeloma activity, while dichloroacetate, an inhibitor of anaerobic glycolysis, can increase the sensitivity of myeloma cells to chemotherapeutic drugs. Pyruvate kinase isoform M2 (PKM2) is also a co-stimulator of hypoxia-inducible factor 1α (HIF-1α) activity and inducer of cPD-L1 inducer in several immune cells. TEPP-46, a PKM2 inhibitor, can down-regulate PD-L1 expression in myeloma cells and neighbouring cells favouring the rescue of Vγ9Vδ2 T cells; B. TCA cycle and mitochondrial metabolism (light green): The TCA inhibitors phosphonoethyl ester of succinyl phosphonate (an α-ketoglutarate dehydrogenase - α-KGDH - inhibitor) and AGI-6780 (an isocitrate dehydrogenase – IDH - inhibitor), the glutaminolysis inhibitor BPTES, and the electron transport chain (ETC) inhibitor α-tocopheryl succinate (α-TOS) are FDA-approved agents under clinical evaluation in several tumors and potentially applicable also to MM. Drugs that induce mitochondria destabilization have emerged as good target for anti-cancer drugs. In particular, retrieving the activity of PGC1α, a transcription factor that promotes mitochondrial biogenesis, may allow to improve the defective mitochondrial mass of immune effector cells in TME, including Vγ9Vδ2 T cells, promoting their activation and cytotoxic functions. C. Hypoxia (grey): The hypoxic TME in MM BM niche leads to the up-regulation of HIF-1α, a strong transcriptional inducer of GLUT and glycolytic enzymes. The inhibition of HIF-1α synthesis or nuclear translocation may be an effective way to reduce glycolytic fluxes in myeloma cells to make glucose available to immune effector cells, including Vγ9Vδ2 T cells. The electron transport chain (ETC) in myeloma cells contribute to hypoxia in the TME and ETC inhibitors can improve the efficacy of ICP/ICP-L blockade by reducing hypoxia in the BM of MM patients. D. Mev pathway (orange): Inhibition of acetyl citrate lyase (ACLY) and acetyl-CoA acetyltransferase-1 (ACAT1) enhances the pro-immunogenic effects of chemotherapy and the efficacy of the antitumor T cells in preclinical models. The Mev pathway can be targeted by HMGCR inhibitors, like statins, or by nitrogen containing bisphophonates (N-BP), like zoledronic acid (ZA). By inhibiting farnesyl pyrophosphate synthase (FPPS), ZA induces the intracellular accumulation of isopentenyl pyrophosphate (IPP) which is recognized by Vγ9Vδ2 T cells. Both statins and N-BP have also direct antitumor effects, inducing apoptosis and inhibiting proliferation of MM. E. Fatty acid metabolism (dark blue): The fatty acid synthase (FAS) and the fatty acid oxidation (FAO) may support respectively the proliferation and differentiation of effector and memory cells, including Vγ9Vδ2 T cells. Long-lasting memory-like T cells rely preferentially on mitochondrial OXPHOS for their energetic demands. Fenofibrate, a PPAR-α activator that increases FAO, sustains the immune competence of CD8+ TIL preventing their functional exhaustion and synergizes with anti-PD-1 blockade. Similarly, chemical agents enhancing OXPHOS in CD8+ TIL enhanced the efficacy of anti-PD1 treatments in solid tumors. Etomoxir has been used to deprive myeloma cells of FA via inhibition of carnitine palimtoyl transferase 1 (CPT1), the pace-maker enzyme in FAO, yielding to a significant suppression of myeloma cell proliferation. The combination of etomoxir with orlistat (a FAS inhibitor) synergistically suppresses myeloma cell growth and enhances sensitivity to bortezomib. F. Amino acid metabolism (pink): IDO inhibitors can rescue dysfunctional or exhausted T cells by restoring tryptophan levels in the TME. Arginine metabolism can be modulated in the TME both by Arg1 inhibition and by targeting its conversion into citrulline by ADI-PEG20. Also PDE5 inhibitors down-regulate Arg1 and nitric oxide synthase–2 (NOS2) expression. Glutamine metabolism can be targeted by glutamine analogs, inhibitors of glutamine transporters or inhibitors of glutamate conversion to α-ketoglutarate. G. Ado pathway (dark green): the ecto-enzymes CD73, CD39, and CD38, and Ado receptors (A2AR) are druggable MCP. A2AR inhibitors, anti-CD73 mAb, anti-CD39 mAb and anti-CD38 mAb therapy demonstrated antitumor activity both in monotherapy as well as in combination with PD-1/PD-L1 blockade.
Glucose consumption is one MCP granting cancer cell survival, restricting T-cell activation and promoting resistance to adoptive T-cell therapy (96). Both anaerobic and aerobic glycolysis are amenable to therapeutic intervention. Myeloma cells exhibit increased activity in glucose transporters of GLUT family like GLUT1, GLUT4, GLUT8 and GLUT11, and in glycolytic enzymes like HK (97). Interestingly, myeloma cells rely on the insulin-responsive glucose transporter GLUT4 for basal glucose consumption, maintenance of Mcl-1 expression, growth, and survival. The FDA-approved HIV protease inhibitor ritonavir acts as a GLUT4 off-target inhibitor abrogating both glucose uptake and proliferation on KMS11 and L363 myeloma cells (97). Interestingly, some myeloma cells can survive to glucose deprivation and/or ritonavir treatment because they switch to OXPHOS to support their energetic needs. Metformin is an FDA approved antidiabetic medication that targets mitochondrial complex 1. The combination of ritonavir and metformin in vitro induces the apoptosis of myeloma cell lines and human primary myeloma cells, and the same combination has shown antimyeloma activity in vivo in a xenograft myeloma mouse model (98). Inhibition of AKT and mTORC1 phosphorylation in combination with Mcl-1 down-regulation have been advocated as key mechanisms (98), but it is also possible that a major contribution comes from the energetic crash induced by the ritonavir and metformin combination. Dichloroacetate, an inhibitor of anaerobic glycolysis, has been reported to increase myeloma cell sensitivity to bortezomib (99), and Zub et al. have confirmed that inhibition of aerobic glycolysis increases sensitivity of myeloma cells to melphalan (100).
Pyruvate kinase isoform M2 (PKM2) catalyzes the final rate-limiting glycolytic reaction and plays a key role in cancer cell metabolism and immune responses. Furthermore, PKM2 dimers can translocate into the nucleus and stimulate HIF-1α-dependent transcriptional activity. Intriguingly, PD-L1 is up-regulated by PKM2 and HIF-1α in macrophages, DC, T cells and tumor cells. The small molecule TEPP-46 partially relieves the immune suppressive TME contexture by down-regulating PD-L1 expression via PKM2 inhibition (101). Additional ICP/ICP-L blockade (i.e., TIM3, LAG-3 etc) can further improve TEPP-46 activity and ameliorate antitumor immune responses.
ICP/ICP-L blockade itself can down-regulate glycolytic fluxes. Anti- ICP/ICP-L mAbs in combination with glycolytic inhibitors can restrain the extensive glucose uptake and utilization by tumor cells and redirect this energy resource towards effector T cells to sustain their reactivity and expansion (94). The increased glycolytic flux in tumors is controlled by AKT/mTOR pathway that up-regulates the glucose uptake via specific GLUT transporters and glycolytic enzymes (67). Anti-PD-L1 mAbs reduce glycolysis in cancer cells by inhibiting Akt/mTOR signaling making glucose also available to T cells to fuel their metabolic needs and mount antitumor immune responses (67, 102, 94). A plus for ICP/ICP-L mAbs compared with traditional glycolytic inhibitors (i.e, fasentin, lonidamine, phosphonoacetohydroxamate, galloflavin, etc) is that they target more specifically tumor cells. The combination of ICP/ICP-L inhibitors with sub-therapeutic concentrations of glycolytic inhibitors (to prevent side effects) can have synergistic effect on the antitumor activity of T cells in the TME.
Akt/mTOR inhibitors also can be combined with ICP/ICP-L mAbs to promote a fairer glucose redistribution in the TME. Interestingly, it has been reported a tight association between ICP-L expression and mTOR activity in cancer cells (103) suggesting that their concurrent inhibition can result in two therapeutic effects: 1) inhibition of tumor cell growth by interfering with the pro-survival pathways driven by Akt/mTOR signaling; 2) suppression of glucose over-utilization by tumor cells and recovery of antitumor immune functions, including Vγ9Vδ2 T cells. Lastly, the combination of ICP/ICP-L blockade and Akt/mTOR inhibition could also increase the sensitivity of myeloma cells to conventional chemotherapy opening the way to triple combination treatments.
The TCA cycle and OXPHOS are mitochondrial pathways acting downstream to aerobic glycolysis. The TCA cycle benefits also from the anaplerotic fluxes of FAO and glutaminolysis. These pathways are able to provide long-term energy supply and provide building blocks for proliferating cells and immune cells undergoing functional differentiation.
Myeloma cells rely on the TCA cycle, including its anaplerotic fluxes, and OXPHOS to meet their energy requirements (14, 15, 104). Disruption of these energy sources has a strong impact on tumor cell survival and proliferation. Mitochondria have emerged as an intriguing target for anticancer drugs, inherent to the great majority of tumors. Drugs that target mitochondria and exert anticancer activity have been termed 'mitocans' to outline their ability to induce mitochondria destabilization (105). The TCA inhibitors phosphonoethyl ester of succinyl phosphonate (an α-ketoglutarate dehydrogenase inhibitor) and AGI-6780 (an isocitrate dehydrogenase – IDH - inhibitor), the glutaminolysis inhibitor BPTES, and the electron transport chain (ETC) inhibitor α-tocopheryl succinate (complex II inhibitor) are FDA-approved agents under clinical evaluation in several tumors and potentially transferable to MM. Targeting the mitochondrial metabolism has also already been explored in MM patients in combination with proteasome inhibitors. The combination of carfilzomib and the IDH2 inhibitor AGI-6780 is lethal to primary myeloma cells in vitro because the TCA cycle is disrupted and ATP is no more available. Since IDH2 is activated by the NAD+-dependent deacetylation mediated by sirtuin-3, the use of NAD+-generating enzyme nicotinamide phosphoribosyltransferase (NAMPT) inhibitors is another potential tool to improve the efficacy of carfilzomib and overcome the development of carfilzomib resistance (106).
Mitocans are not exempted from detrimental effects because mitochondrial biogenesis is also required by effector T cells to mount effective antitumor immune responses. Tumor-infiltrating lymphocytes (TIL) are characterized by low mitochondrial mass and metabolic flux due to the Akt-mediated inhibition of peroxisome proliferator-activated receptor gamma coactivator 1α (PGC1α), a transcription factor that promotes mitochondrial biogenesis. Restoring the activity of PGC1α corrects the defective mitochondrial mass and increases the energy supply for TIL, promoting their sustained activation and effector functions (107). Mitochondrial biogenesis is emerging as critical issue also to improve the efficacy and duration of chimeric antigen receptor (CAR) redirected T cells (CAR-T cells). The costimulatory domain 4-1BB has replaced CD28 in the architecture of second-generation CAR-T cells because it promotes the outgrowth of CD8+ memory T cells, which have significantly better respiratory capacity, increased FAO and enhanced mitochondrial biogenesis (108). Very recently, Van Bruggen et al. have reported that the clinical efficacy of CAR-T cells in CLL is dependent on their mitochondrial mass which can be regarded as an indirect evidence of their capacity to successfully compete with energy sources in the TME (109).
Unfortunately, mitocans, like other metabolic correctors, are penalized by the lack of specificity leading to contrasting effects on tumor cells and immune effector cells in the TME. It is a matter of debate whether the conjugation of mitocans with tumor-selective ligands and/or the conjugation of mitochondrial reactivating agents (e.g. PGC1α inducers) with T-lymphocytes-targeting ligands may overcome this impasse. Conjugation of mitochondrial activators with pAgs specifically recognized by Vγ9Vδ2 T cells may represent a promising strategy to improve the selectivity of metabolic correctors.
The hypoxic TME in MM BM leads to the constitutive up-regulation of HIF-1α, a strong transcriptional inducer of GLUT and glycolytic enzymes. Therefore, HIF-1α inhibition can decrease glucose utilization by myeloma cells making glucose available to immune effector cells including Vγ9Vδ2 T cells. Unfortunately, HIF-1 inhibitors have failed clinical expectations so far, probably because they lack specificity (110). Oxygen consumption by ETC in tumor cells is also under investigation as a targetable MCP. Hypoxia promotes ICP/ICP-L expression via HIF-1α activation and other mechanisms and preliminary data indicate that ETC inhibitors attenuates hypoxia in the BM of MM patients and increase the efficacy of anti- ICP/ICP-L mAbs (107).
These data confirm that the ICP/ICP-L circuitry is an attractive candidate to combine with HIF-1 inhibitors. Hypoxia increases the number of PD-L1+ MDSC and Tregs in the TME, and targeting hypoxia-driven MCP can prevent the accumulation of these immune suppressive cells (111). The interactions between hypoxia-driven MCP and ICP/ICP-L can also be mediated by exosomes. Interestingly, it has been reported in oral squamous cell carcinoma that exosomes derived from normoxic tumors expand immunocompetent γδ T cells (mainly represented by Vγ9Vδ2 T cells), whereas exosomes derived from hypoxic tumors expand PD-L1+ MDSC that recruit PD1+ anergic γδ T cells (112). This work demonstrates that oxygen pressure in the TME regulates the induction of anti- or pro-tumoral γδ T cells by modulating the release of exosomes by tumor cells.
HIF-1β, a transcription factor steadily translocated in the nucleus to activate the transcriptional program mediated by HIF-1α in hypoxia, is a strong inducer of ABCA1 which is highly expressed in hypoxic endothelial cells (113) and macrophages (114). BMSC of MM patients, deep-rooted in a constitutively hypoxic TME, display high levels of ABCA1 and extrude huge IPP amounts in the TME (49). We are currently investigating whether these unnatural IPP concentrations (in the micromolar range) determine a functional exhaustion of Vγ9Vδ2 T cells by increasing, in the absence of appropriate co-stimulatory signals, the expression of multiple ICP (PD-1, TIM3, LAG-3) (47, 5, 49, 7). We have initiated to investigate whether HIF-1 inhibitors can rescue the immune competence of Vγ9Vδ2 T cells in the TME of MM patients by decreasing ABCA1 expression and extracellular IPP release from BMSC. As said above, metabolic corrections are often double-edged approaches. In this case, decreasing the HIF-1/ABCA1 axis in BMSC may limit the long-term and uncontrolled exposure of BM Vγ9Vδ2 T cells to supra-physiological IPP concentrations and relieve their anergy and senescence, but the down-regulation of IPP production and release by DC may concurrently restrain the activation of Vγ9Vδ2 T cells. In-depth investigation of different expression of cell surface antigens by BMSC vs DC, coupled with medicinal chemistry approaches to conjugate HIF-1 inhibitors with ligands specific for immune suppressive cells, are current under investigation to successfully introduce HIF-1 inhibitors in the clinical setting.
Interestingly, metformin, an anti-diabetic drug that inhibits OXPHO, reduces oxygen consumption in the TME and improves oxygen concentrations (115, 116). The reduced hypoxia relieves several immune suppressive features and increases the efficacy of PD-1 blockade. Metformin alone does not have a significant antitumor activity, but in combination with anti-PD-1 mAb locally increases the number and function of CD8+ cells with antitumor activity (i.e. producing TNF-α and IFN-γ) (107). It is currently unknown the impact of metformin or other hypoxia-targeted treatments on Vγ9Vδ2 T cells under normoxic or hypoxic conditions. Nevertheless, since Vγ9Vδ2 T cells are highly sensitive to the metabolic TME alterations, including hypoxia, it is very likely that remodeling the hypoxic tone in the TME of MM patients is immunologically beneficial also to Vγ9Vδ2 T cells. Besides relieving hypoxia, metformin can be particularly useful in MM patients because it consistently decreases IL-6R expression in myeloma cells (117). IL-6 can promote, in combination with TGF-β and other cytokines highly abundant in the TME of MM patients, the differentiation of IL-17 producing γδ T cells which support tumor cell progression by promoting angiogenesis (118).
Lastly, hypoxia is considered a strong protumoral metabolic change in the TME because it favours the emergence of MDR (119). However, recent data suggest that hypoxia can be exploited to induce the selective activation of prodrugs in the TME. For instance, evofosfamide is a prodrug of the DNA alkylator bromo-isophosphoramide that is activated under hypoxic conditions. environment. Evofosfamide in combination with bortezomib and dexamethasone has shown a safe profile and clinical efficacy in patients with relapsed MM who have failed standard treatments (120).
ICP/ICP-L blockade can also be combined with Mev pathway manipulation to improve antitumor immune responses. Promising data have been generated in murine cancer models where inhibition of acetyl citrate lyase (ACLY), the main generator of cytosolic acetylCoA used in cholesterol synthesis, enhances the pro-immunogenic effects of chemotherapy and the efficacy of antitumor T cells. The inhibition of acetylCoA acetyltransferase-1 (ACAT1) that promotes a feedback inhibition of Mev pathway by preventing the esterification of cholesterol, produces similar effects (121, 122). These two metabolic modifications can be used in association with ICP/ICP-L blockade to boost the antimyeloma activity of Vγ9Vδ2 T cells.
Modulation of the Mev pathway can take advantage of the hypoxic TME of MM patients. HMGCR is down-regulated by hypoxia. Direct inhibitors of HMGCR like statins or down-stream FPPS inhibitors like NBP are more cytotoxic against cancer cell lines under hypoxic than normoxic conditions (123). One possible explanation is that the deeper Mev pathway inhibition leads to a drastic intracellular deprivation of cholesterol and isoprenoids that myeloma cells cannot tolerate under hypoxic conditions. Another mechanism can be the BTN3A1 conformational changes induced by NBP-induced intracellular IPP accumulation cells that can increase myeloma cell recognition by Vγ9Vδ2 T cells. The first mechanism may explain why statins and compounds like brutieridin and melitidin, that incorportate the HMG moiety (124), may have potent antitumor effects (125-127) even if they do not induce IPP accumulation and stimulate Vγ9Vδ2 T cells (5, 47, 128). Since lipohilic statins inhibit HIF-1α activity in solid tumors, it is worth investigating whether these drugs can also suppress PD-1L transcription. So far, no data are available about the capacity of statins to inhibit HIF-1α activation in tumor-infiltrating lymphocytes and/or Vγ9Vδ2 T cells. Future research in this field may help to understand whether the combination of lipophilic statins and ICP/ICP-L blockade may enhance antitumor immune responses in the TME of MM patients and eliminate residual MDR+ myeloma cells nested in the hypoxic BM niches.
Beside the Mev pathway, other lipid metabolic pathways are important mediators of antitumor immune responses and therefore potentially druggable MCP. Any modifications in cellular lipid metabolism significantly affects T-cell fate and function (129). Indeed, the activation-induced proliferation and differentiation of effector T cells is supported by FAS, whereas the development of CD8+ T cell memory cells requires FAO (130, 131). As a consequence, long-lasting memory-like T-cells rely preferentially on mitochondrial OXPHOS and FAO for meeting their energetic demands (130, 132).
Enhanced FA utilization can be an important metabolic opportunity for exhausted T cells that are highly dependent on FAO to support their energetic needs. In the presence of glucose deprivation, FAO becomes the main fueling pathway to preserve the effector functions of CD8+ T cells in the TME. Fenofibrate, a PPAR-α activator with the capacity to increase FAO, sustains the immune competence of CD8+ TIL and delays their functional exhaustion irrespective of PD-1 expression. Interestingly, fenofibrate has been reported to synergize with anti-PD-1 blockade in melanoma (133). Similarly, chemical agents enhancing OXPHOS in CD8+ TIL increase the efficacy of anti-PD1 treatments in colon cancer models (134). These data suggest that increasing FAO and OXPHOS in TIL, in combination with anti-ICP/ICP-L treatment, can represent a significant advance in preventing the functional exhaustion of TIL in the TME. Unfortunately, increasing FAO in T cells may induce unwanted effects on the delicate balance between activation and inhibition of immune cells. FAO is also the main energetic pathway used by Treg cells and MDSC (135-137) to differentiate and exert their immune suppressive activity on T cells. As a consequence, FAO inhibition can prevent Treg accumulation, but it impairs effector T-cell functions; contrariwise, FAO up-regulation can improve T-cell functions, but it increases immunosuppressive functions by Treg cells and MDSC (135).
Lipid metabolism can play a special role in BM-located malignancies like MM. The proportion of BM adipocytes increases with age and therefore it is not surprising that these cells are particularly abundant in the BM of MM patients which is a disease of the older population. Adipocytes in the TME of MM patients can provide FA and triglycerides to myeloma cells (138) that are strategically equipped with appropriate receptors like CD36 and the FA Transporter Proteins (FATPs) to internalize these invaluable energy sources (139, 140). Etomoxir has been used to deprive myeloma cells of FA via inhibition of carnitine palimtoyl transferase 1 (CPT1), the pace-maker enzyme in FAO, yielding to a significant suppression of myeloma cell proliferation (140). Myeloma cells can also produce FA via FAS and this pathway is instrumental to metabolically support their survival and expansion. Interestingly, the combination of etomoxir with orlistat (a FAS inhibitor) synergistically suppresses myeloma cell growth and enhances the sensitivity to bortezomib treatment (141). Although promising, these findings should be regarded with caution because the results have been generated using myeloma cell cultures taken out of the immune suppressive TME.
Very little data are available about lipid metabolism in Vγ9Vδ2 T cells. Recently, it has been shown that IL-21 reduces aerobic glycolysis and increases FAO in T cells. This metabolic reset is paralleled by increased mitochondrial biogenesis to generate long-lasting memory T cells with low PD-1 expression (142). Interestingly, normal Vγ9Vδ2 T cells have enhanced antitumor activity if expanded in the presence of IL-21 and IL-2, whereas Vγ9Vδ2 T cells from cancer patients have defective reactivity to IL-21 stimulation (143). Vγ9Vδ2 T cells from patients with acute myeloid leukemia display lower expression of IL-21R and require higher levels of IL-21 for their expansion. These cells also express significantly higher TIM-3 levels than healthy Vγ9Vδ2 T cells and up-regulate TIM-3 after IL-21 stimulation. Blocking TIM-3 increases the proliferation and reactivity of Vγ9Vδ2 T cells to IL-21 stimulation (144). These data indicate the existence of druggable interconnections between ICP/ICP-L and FA metabolism mediated by cytokines like IL-21. Preliminary data in our laboratory indicate that anergic Vγ9Vδ2 T cells isolated from the TME of MM patients may up-regulate multiple ICP when stimulate by ZA-treated DC in the presence of IL-2. Altogether, these data confirm the multifaceted reactivity of Vγ9Vδ2 T cells embedded in the TME and how their functional plasticity dangerously allows them to switch from antitumoral to protumoral functions (145).
IDO inhibition is under investigation as a druggable MCP to improve the efficacy of ICP/ICP-L inhibition. IDO inhibitors can rescue dysfunctional or exhausted T cells by raising tryptophan levels in the TME. L-methyl-tryptophan (1-MT) (146) and INCB024360 (147) have been shown to improve the immune responses of tumor-specific T cells in mouse models. Following these initial observations, indoximod (IDO1 and IDO2 inhibitor), navoximod (IDO1 inhibitor, also known as GDC-0919 and NLG919) and the dual IDO1–TDO inhibitors HTI-1090 (IDO1-TDO inhibitor) also known as SHR9146) and DN1406131 have been developed and investigated as single agents or in combination with ICP/ICP-L blockade (148). Holmgaard et al. have shown that IDO inhibition in combination with CTLA-4, PD-1/PD-L1, and GITR blockade has synergistic activity on tumor cell growth and improves the survival in different tumor models (149). Based on these promising results, clinical trials are currently underway to investigate the combination of anti–CTLA-4 mAb and IDO inhibition in patients with melanoma. The very potent and highly selective IDO1 inhibitor epacadostat (INCB024360) is under clinical investigation as single agent in various advanced-stage malignancies and under consideration in combination with anti-ICP/ICP-L mAbs.
The combination of IDO inhibitors and ICP/ICP-L blockade is also worth investigating in in MM given the well-known role played by in the TME of these patients. A preclinical rationale has been provided by An et al. who have shown that anti-PD-L1 mAb in combination with IDO inhibition can overcome the osteoclast-induced suppression of T-cell mediated antimyeloma immune responses (150). IDO inhibition can also alleviate Vγ9Vδ2 T-cell immunosuppression induced by the overproduction of kynurenines by myeloma cells and MDSC in the TME.
Arginine deprivation is another major MCP restraining antitumor immune responses mediated by T cells and NK cells in the TME (151). Arginine supply and/or prevention of arginine degradation via Arg1 inhibition are feasible strategies to reinstate antitumor immune responses in the TME. As expected, arginine supplementation revives the cytotoxic functions of T cells and NK cells, and, in combination with anti-PD-L1 mAb, enhances antitumor immune responses and prolongs the survival of tumor-bearing mice (152). CB-1158, a potent and orally-bioavailable small-molecule Arg1 inhibitor, has shown efficacy in murine syngeneic tumor models (153) as single agent or in triple combination with adoptive T-cell or NK-cell therapy, adoptive NK cell therapy, and anti-PD-L1 mAb. The combination of CB-1158 and anti-PD-L1 mAb is currently under clinical investigation in patients with advanced/metastatic solid tumors (NCT02903914, INCB 01158-10) (154).
Interestingly, arginine metabolism can also be modulated in the TME by targeting its conversion into citrulline. In the presence of arginine deprivation, normal tissues can increase citrulline uptake and up-regulate the expression of argininosuccinate synthetase 1 (ASS1) which converts citrulline into arginine. This auxotrophy mechanism can be defective in tumor cells that are unable to up-regulate or lack ASS1 (155). Pegylated arginine deiminase (ADI-PEG 20) has been develop to convert arginine into citrulline and ammonia, making ASS1-deficient tumor cells unable to use citrulline to generate arginine and meet their metabolic needs (156). ADI-PEG 20 has shown antitumor activity in ASS1-deficient tumor cells from patients with breast cancer (157), small-cell lung cancer (158) and acute myeloid leukemia (159). Interestingly, T cells can increase citrulline uptake and up-regulate ASS1 expression under low arginine conditions (160-162). Thus, the conversion of arginine into citrulline by ADI-PEG 20 may have different effects on T cells compared with arginine depletion induced by Arg1. Indeed, Brin et al. have shown that peripheral blood T cells from healthy donors can be fully activated in the presence of ADI-PEG 20 avoiding PD-1 up-regulation and the differentiation into Treg cells. In murine syngeneic tumor models, ADI-PEG 20 has been reported to promote the recruitment of T cells in the TME and improve the efficacy of anti-PD-L1 treatment (163).
Targeting Arg1 can also be effective in MM. We have shown that PD-L1+ MDSC are significantly expanded in the TME of MGUS and MM patients, irrespective of the percentage of TME-infiltrating myeloma cells (5). Romano A. et al. have recently shown that Arg-1 is highly expressed by MDSC of MM patients (164). Arg-1 inhibition improves the in vitro antimyeloma activity of bortezomib and lenalidomide and this combination is currently tested in animal models to determine safety and efficacy in vivo. We are currently investigating whether Arg-1 up-regulation and arginine deprivation are involved in the dysfunction of BM Vγ9Vδ2 T cells in MGUS and MM patients.
Borrello’s group has pursued a different strategy to target the immune suppressive functions of MDSC in the TME of MM patient (165). They have shown that sildenafil down-regulates Arg1 and nitric oxide synthase–2 (NOS2) expression, thereby reducing the suppressor function of MDSC and ameliorating antimyeloma immune responses mediated by T cells. In the clinical setting, they have shown that tadalafil, another phosphodiesterase-5 (PDE5) inhibitor, attenuates the suppressor functions of MDSC and allows the achievement of measurable antimyeloma immune responses (166). When added to chemotherapy, this approach has improved the clinical outcome of end-stage relapsed/refractory MM. We have investigated the effects of PDE5 inhibitor sildenafil on pAg-reactivity of BM Vγ9Vδ2 T cells from MM patients and failed to show any functional recovery, further confirming the deeper impairment of these cells compared with conventional T cells (5).
MDSC also express high levels of NOS2, which convert arginine into NO. NO can suppresses T-cell function either by S‑nitrosylation of critical cysteine protein residues or by regulation of guanylyl cyclase and cyclic GMP-dependent kinases (167). Sharing the same substrate, NOS2 and Arg-1 are in dynamic competition with the former having much higher affinity for arginine, but much slower catalytic activity. In the presence of severe arginine depletion, NOS2 switches from arginine degradation to the generation of reactive oxygen species (ROS) and reactive nitrogen species which are immune suppressor metabolites. These data suggest that the combination of Arg1 and NOS2 inhibitors should ensure the most effective MDSC inhibition. NCX‑4016 (nitroaspirin), a small molecule developed to test this hypothesis, has been shown to correct the immune dysfunction and promotes tumor eradication after cancer vaccination in tumor-bearing mice (168).
Targeting NOS2 could be particularly rewarding to rescue the immunocompetence of Vγ9Vδ2 T cells. Douguet et al. have shown that a significant proportion of γδ T cells infiltrating mouse and human melanoma express NOS2. Experimental data in the murine melanoma model indicate that NOS2 can be involved in the generation and expansion of IL-17-producers tumor-promoting γδ T cells which, in turn, can promote the recruitment and accumulation of MDSC in the TME (169). It is currently unknown whether NOS2 is also expressed by BM Vγ9Vδ2 T cells from other cancers, but the combination of NOS2 inhibitors and ICP/ICP-L blockade sounds very interesting to recruit immune effector cells, including Vγ9Vδ2 T cells, in the TME of MGUS and MM patients.
Glutamine metabolism is critical for tumor cell survival and therefore another attractive MCP to target for therapeutic interventions. The whole pathway has been considered and several compounds developed, from glutamine analogs to inhibitors of glutamine transporters or inhibitors of glutamate conversion to α‑ketoglutarate (170). Some of these compounds have moved on to clinical trials as single agents or in association with anti-ICP/ICP-L mAbs (148).
The importance of glutamine metabolism in myeloma cells have arisen from the clinical observation that a significant proportion of MM patients at diagnosis are affected by hyperammonemia and encephalopathy in the absence of liver dysfunction. Subsequently, it has been shown that human myeloma cell lines and primary myeloma cells, lacking a detectable expression of glutamine synthetase (GS), rely on the uptake of extracellular glutamine, mainly via the ASCT2 glutamine transporter, for their growth and survival. Bolzoni et al have shown that blocking glutamine uptake significantly affects the viability of myeloma cells and their sensitivity to bortezomib and other drugs. They have also shown that stable ASCT2 downregulation by a lentiviral approach inhibits the growth of human myeloma cell in vitro and in a murine model. Similar data have been reported using the combination of glutaminase (GLS) inhibitor CB-839 alone or in combination with pomalidomide (171, 172) or glutamine deprivation in the presence of venetoclax (173).
Targeting glutamine metabolism in myeloma cells may have many-sided effects on T cells in the TME. Glutamine uptake and glutaminolysis are up-regulated in naïve T cells after activation to support their proliferation (62). Following activation, glutamine levels can regulate the functional differentiation of specific CD4+ and CD8+ subsets. In the presence of adequate glutamine supply, effector T cells can shift from glycolysis to OXPHOS to generate central memory-like cells with enhanced survival and antitumor activity (71). In the presence of glutamine deprivation, including ASCT2 deletion, T-cell activation results in the differentiation into Foxp3+ Tregs cells that further suppress T-cell activity (70). More recently, it has been shown that inhibition of glutaminolysis at the level of GLS can have different effects on T-cell differentiation compared to depletion of extracellular sources. Johnson et al have shown that GLS promotes the functional differentiation of Th17 cells in spite of Th1 cells. Transient GLS inhibition leads to increased Th1 differentiation and the generation of CD8+ CTL effector functions (174). It is currently unknown whether glutamine metabolism, either as deprivation of external source or as impaired GLS activity, may impact on the functional differentiation of Vγ9Vδ2 T-cell subsets. Interestingly, IL-17-producing γδ T cells have been reported to be generated in the TME of tumor-bearing mouse and to behave as tumor-promoting cells (175). We are currently investigating whether the glutamine deprivation induced by myeloma cells in the TME in the presence of IL-6 and TGF-β promotes the generation of IL-17-producing Vγ9Vδ2 T cells as in tumor-bearing mice.
Given the strong impact that manipulation of glutamine metabolism may have on antitumor immune responses, it is not suprising that clinical trials are under investigation in combination with anti-ICP/ICP-L mAbs. Trigriluzole (BHV-4157), a compound that reduces extracellular glutamate levels, is currently tested in combination with nivolumab or pembrolizumab in metastatic or unresectable solid tumors or lymphoma (NCT03229278), while CB-839 (glutaminase 1 inhibitor) is tested in combination with nivolumab in advanced- stage clear cell renal carcinoma, melanoma and non small cell lung cancer (NCT02771626) (148).
Ado is an important MCP in the hypoxic BM niche where residual myeloma cells can survive chemotherapy and immunotherapy (176). Ado, through its receptor A2AR, can up-regulate PD-1 expression on CD8+ and Treg cells (177) and the dual blockade of PD-1 and A2AR synergistically increases the cytotoxic potential of CD8+ cells and controls tumor growth (178). Combination of A2AR inhibition with TIM-3 or CTLA-4 blockade has also been shown to be effective in enhancing antitumor immune responses in a variety of syngeneic tumor models (179).
The role of Ado in the immune suppressive TME of MM patients is also under investigation (180). Yang R et al. have reported increased Ado concentration in the BM plasma from MM patients compared with healthy controls (181). CD39 expression by myeloma cells coupled with CD73 expression by BMSC and other bystander cells are responsible for the generation of immunosuppressive extracellular Ado concentrations in the TME leading to A2AR-mediated T-cell inhibition. The CD39 inhibitor POM1 inhibits Ado generation in myeloma cell/BMSC co-cultures and restores T-cell proliferation in vitro. Interestingly, high CD39 expression in the TME of MM patients confers a poor clinical outcome further supporting the role Ado in the immunosuppressive TME of these patients (181). Richles RJ et al. have shown that A2AR agonists in combination with PDE inhibitors act synergistically in triple combination with glucocorticoid to suppress the proliferation of myeloma cells in vitro (182).
Extracellular Ado in the TME of MM patients can also be generated from nicotinamide dinucleotide (NAD) by the concerted action of CD38, CD203a and CD73 (183). CD38 is a multifunctional ecto-enzyme acting also as a receptor and adhesion molecule in the TME of MM patients. Being expressed by myeloma cells and several other lymphoid and myeloid cells in the TME, CD38 is strategically located as a target intersection to modulate tumor-host interactions (184). Several anti-CD38 mAb like daratumumab, isatuximab, and MOR202 have been developed and clinically used in MM patients as single agents or in combination with steroids and other drugs (185). Although several cell subsets express both CD38 and ICP/ICP-L in the TME of MM patients (including myeloma cells, immune suppressor cells and immune effector cells), very little data are available about the cascade of reciprocal interactions eventually triggered by anti-CD38 or anti-ICP/ICP-L mAbs. Chen et al. have reported in lung cancer and animal models that acquired resistance to anti-PD-1/PD-L1 mAbs is mediated by CD38 upregulation and shown that concurrent CD38 and PD-1/PD-L1 inhibition improves antitumor immune responses (186).
The role of Ado pathway in γδ T-cell functions is much less recognized. Liang D. et al. have addressed this issue in an experimental autoimmune uveitis mouse model (187). They have shown that the CD73 and A2AR expression on γδ T cells are dependent on their activation status and modulate immune responses mediated by Th17 and Treg cells by fine-tuning extracellular Ado concentration (188, 189). CD73 and CD39 expression has been correlated with the acquisition of suppressor functions in Vγ9Vδ2 T cells after exposure to pAg in the presence of inappropriate cytokines (190, 191). CD39 upregulation has been reported to dephosphorylate self and microbial pAgs restraining the reactivity of Vγ9Vδ2 T cells (192).
The TME of MM patients is affected by a metabolic reset orchestrated by myeloma cells when their number exceeds the physiological threshold (2-3%) that the BM is well-suited to accommodate. The metabolic reset is finalized to sustain myeloma cell growth and promote resistance to chemotherapy and immunosurveillance. Hypoxia, low pH, nutrients deprivation, and toxic waste compromise the antimyeloma activity of immune effector cells (CD8+ cells, NK cells, Vγ9Vδ2 T cells, etc) and drive the differentiation of immune suppressor cells (Tregs, MDSC, M2 macrophages, etc). Vγ9Vδ2 T cells are among the first immune effector cells to fall prey to the immune suppressive TME elaborated by myeloma cells. The expression of ICP/ICP-L is facilitated by metabolic alterations and this deadly network makes very challenging to rescue the immunocompetence of Vγ9Vδ2 T cells and other immune effector cells.
A growing body of evidence indicates that targeting MCP in combination with ICP/ICP-L blockade is a strategy that can significantly improve the efficacy of immunotherapy. This strategy can be applied ex-vivo using metabolic correctors in adoptive immunotherapy trials to make immune cells more resistant to the metabolic challenges encountered upon reinfusion. Alternatively, metabolic correctors could be used in vivo in association with ICP/ICP-L blockade in the remission phase when most of myeloma cells have been eliminated by chemotherapy. This approach is feasible in disease like MM where current treatments based on the combination of autologous stem cell transplantation (ASCT) with novel drugs can achieve complete remission with undetectable minimal residual disease in a significant proportion of patients. We have shown that the immune suppressive TME imprinting is not reversed in MM who are in complete remission after ASCT. This can represent a privileged setting for immune interventions because metabolic correctors and ICP/ICP-L blockade are expected to target mainly immune cells and, in the absence of myeloma cells, to restore effective and long-lasting immunosurveillance.
This study was supported by the Italian Association for Cancer Research (AIRC) (IG15232 and IG21408 to CR; IG16985 and IG21744 to MM). B.C. is a post-doc research fellow supported by AIRC.