
Small non-coding PIWI-interacting RNAs (piRNAs) silence the expression of transposable elements integrated in a wide range of eukaryotic genomes in germline cells. Additionally, piRNAs regulate chromatin modifications, such as trimethylation of histone H3 lysine 9 (H3K9me3) or DNA methylation. In the past decade, the roles of piRNAs have been characterized in somatic cells, including post-mitotic neurons. More recently, piRNAs have been shown to play important roles in brain functions and various neuronal diseases, including neurodegenerative disorders. In this review, we introduce recent findings showing the potential involvement of piRNAs in the etiology of different neurodegenerative diseases, including amyotrophic lateral sclerosis (ALS), Alzheimer’s disease (AD) and Parkinson’s disease (PD). These pioneering studies on disease-associated small RNAs will contribute to improving our understanding of the pathogenesis of these neurodegenerative diseases and the development of novel therapeutic strategies.
PIWI-interacting RNAs (piRNAs) are small non-coding RNAs that suppress transposable elements (TEs) (1.2) and maintain chromatin structures in eukaryotic genomes (3.4.5..6.). Additionally, piRNAs modulate the expression of the genomic loci harboring TEs. TEs, which probably invaded the eukaryotic genomes through virus infections in ancient times (7), are spread by unknown mechanisms. In early embryonic development, new TEs can be integrated in the germline genomes and induce genetic diversity in offspring (8), a process in which piRNAs are involved (9). Although the effects of piRNAs had been thought to be restricted to germline cells, piRNAs have recently been shown to function in somatic cells as well. TEs also function in somatic cells and drive genomic heterogeneity in the brain and other tissues (10.11). On the other hand, TEs are deleteriously activated in aged neurons and may induce neurodegeneration (12). In particular, LINE-1 retrotransposons, which are amplified by “copy-paste transposition”, have been reported to be responsible for neurodegeneration because LINE-1 is widely spread throughout mammalian genomes (13.14). In this context, piRNAs are implicated in neuronal activity and the etiology of neuronal diseases by regulating TE expression.
Here, we summarize the involvement of piRNAs in a variety of neuronal diseases by featuring three key studies. 1) Inactivation of ALS-associated FUS causes aberrant precursor piRNA production, leading to abnormal RNA-protein aggregation with FUS in the cytoplasm. The disease-associated consequences are facilitated by Aubergine (Aub), which belongs to a PIWI subfamily of the argonaute protein (15). 2) Depletion of piRNAs and chromatin decondensation by pathogenic Tau stimulates the ectopic expression of TEs and subsequent aberrant cell cycle transitions, leading to neurodegeneration in subjects with AD (16). 3) Profound dysregulation of RNA expression, including piRNAs, has been observed in post-mitotic neurons derived from patients with PD (17).
The roles of piRNAs in neuronal diseases have been extensively analyzed in humans and a variety of animal models, including Drosophila, potentially contributing to further improving our understanding of the pathogenesis of neuronal diseases, the development of appropriate diagnostic markers and therapeutic targets for these diseases.
Compared with miRNAs, piRNAs are longer: generally 21 to 35 nucleotides (nt). The biogenesis of piRNAs is conserved in different animal cells and mediated by PIWI proteins, with some differences (reviewed in 18.19.20.21.22). In mammalian cells, PIWIL1-4 are involved in piRNA biogenesis (Table 1). In Drosophila, Piwi, Aub and Ago3 function as PIWI proteins (Table 1). The process is well characterized in mice and Drosophila, particularly in germline cells (Figure 1). piRNAs are generated through two different biogenesis pathways: primary and secondary piRNA biogenesis. In primary biogenesis, long precursor piRNAs (pre-piRNAs) are transcribed from piRNA clusters, which are defined as specific genomic regions harboring a number of TEs. These pre-piRNAs are cleaved to generate mature piRNAs (characterized by a uracil in the 5’ end of antisense piRNAs) by a protein complex comprising endonucleases (Zucchini in Drosophila and as yet unidentified enzymes in mice) and PIWI proteins (MIWI and MILI in mouse and Piwi and Aub in Drosophila) in the cytoplasm. In secondary biogenesis, mature piRNAs are amplified through the processing of TE-containing mRNAs and pre-piRNAs by PIWI proteins (MILI and MIWI2 in mouse and Aub and Ago3 in Drosophila), a process known as the ping-pong cycle. In this cycle, antisense piRNAs, which begin with uracil, complementarily bind to RNAs with an adenine at position 10. The double-stranded RNAs are loaded onto PIWI proteins to guide the next round of amplification. These RNA species show a characteristic 10-nucreotide (nt) complementarity, which is referred to as a ping-pong signature. The PIWI (MIWI2 in mouse and Piwi in Drosophila)/piRNA complex is translocated to the nucleus to suppress TE expression in the genomic DNA by inducing chromatin modifications, such as trimethylation of histone H3 lysine 9 (H3K9me3) or DNA methylation. In Drosophila somatic cells, pre-piRNAs are transcribed from soma-specific piRNA clusters such as flamenco, then exported to the cytoplasm and further cleaved by protein complexes containing PIWI (Piwi) to produce mature piRNAs (23.24.25.26.27.28.29.30). Soma-specific piRNAs also contribute to the silencing of genomic regions containing TEs.
 Figure 1.
Figure 1.
piRNA biogenesis in the mouse and Drosophila.In primary piRNA biogenesis, pre-piRNAs are transcribed from piRNA clusters, then exported to the cytoplasm and further cleaved by protein complexes containing PIWI proteins (MIWI and MILI in mouse, Piwi and Aub in Drosophila) and endonucleases to produce mature piRNAs. In secondary piRNA biogenesis, mature piRNAs are loaded onto PIWI proteins (MILI in mouse, Aub in Drosophila) to direct the cleavage of complementary TE-containing mRNAs. The cleaved TE-containing RNAs are then loaded onto another PIWI protein (MIWI2 in mouse, Ago3 in Drosophila) to cleave complementary pre-piRNAs, a process known as the “ping-pong cycle”. Mature piRNAs bind to PIWI (MIWI2 in mouse, Piwi in Drosophila) and are re-imported to the nucleus to induce repressive chromatin modifications in the genomic regions of piRNA clusters containing TEs.
| Species | PIWI proteins | |||
| Human | HIWI (PIWIL1) | HILI (PIWIL2) | HIWI3 (PIWIL3) | HIWI2 (PIWIL4) |
| Mouse | MIWI (PIWIL1) | MILI (PIWIL2) | – | MIWI2 (PIWIL4) |
| Drosophila | Piwi | Aub | – | Ago3 |
In the Drosophila brain, piRNAs, which were initially identified as piRNA-like molecules, were shown to be derived from TE sequences (31). Subsequently, piRNA-like molecules were also detected in the cerebral cortex of rhesus macaque (32). Furthermore, MIWI/piLRNAs and a type of piRNA were identified in the dendritic compartment of cultured mouse hippocampal neurons (33). Interestingly, the same study revealed that piRNAs regulate spine morphogenesis by targeting the coding regions of several neuronal mRNAs associated with spine formation. The Piwi/piRNA complex enhances memory-related synaptic plasticity by facilitating serotonin-dependent methylation of the CREB2 promoter in the Aplysia central nervous system (CNS) (34). Thus, piRNAs affect neurogenesis and synaptic plasticity by regulating gene expression at the transcript and genomic levels.
Long interspersed nuclear elements (LINEs) are transcribed at high levels in various rat tissues, including the brain, and their expression is controlled by DNA methylation and non-coding RNAs, including piRNAs (35). Therefore, piRNAs are suggested to have a role in the tissue-specific variation of genomes through the expression and mobilization of LINEs. The expression of TEs and PIWI proteins, such as Aub and Ago3, shows an inverse correlation in the αβ neurons of the mushroom body in Drosophila (11). Based on these findings, piRNAs regulate genomic heterogeneity in neurons by controlling TE transposition.
Defects in the PIWI protein MILI induce hypomethylation of LINE1 promoter regions and induce behavioral deficits in mice, such as hyperactivity and reduced anxiety (36). These effects are likely caused by the inactivation of piRNA biogenesis, although the precise roles of piRNAs in the CNS remain to be determined.
Altered expression of 105 piRNAs was identified in the cerebral cortex of ischemic rats compared with the sham group (37). Those piRNAs suppress the activation of TEs inserted in a variety of genomic regions. The piRNA promoter regions contain putative binding sites for several transcription factors, suggesting that piRNAs are regulated by transcription factors. The changes in piRNA expression observed upon focal ischemia might influence the activity of TEs and subsequent genomic changes, although researchers have not yet presented evidence that this event contributes to brain damage.
The biogenesis of piRNAs is activated during axon regeneration after injury (38.39). Rett Syndrome is a severe neurodevelopmental disorder, accompanied with intellectual disability in girls. Patients show developmental delay, including hand movements, language and communication. The expression of piRNAs is also increased by the loss of methyl-CpG binding protein 2 (MeCP2), mutations of which are responsible for Rett syndrome, in the murine cerebellum (40). Because piRNAs are expressed from TEs, TE activity and mobility might be increased in this model. Although the analysis was performed without replicated samples and further studies are warranted to confirm the findings, piRNAs represent potential clinical biomarkers for the diagnosis of Rett Syndrome.
Both knockdown and mutations in PIWIL1 induce defects in neuronal migration in the developing rat cerebral cortex (41). PIWIL1 knockdown also affects the multipolar–bipolar transition in post-mitotic neurons, which is essential for proper radial migration (41). Furthermore,PIWIL1 loss decreases the expression of microtubule-associated proteins such as Tau, MAP1B in the cortical tissues of adult mice. A whole exome sequencing study of families with simplex autism spectrum disorder identified strong correlations between de novo mutations in PIWI family members, particularly PIWIL2 and PIWIL4, and autism (42). Thus, PIWI family proteins likely regulate brain development, the dysregulation of which would likely be associated with autism and related disorders.
Gulf War Illness (GWI) is a chronic disorder suffered by 25% of veterans of the United States Army who were deployed to the Persian Gulf during the 1990–1991 Gulf War. Memory dysfunction and depression have been reported as the most prominent symptoms in brain and reduced volumes of cortical white and gray matters have also been reported (43.44). While the etiology of GWI is not completely understood, the ingestion of pyridostigmine bromide as a drug to prevent lethal nerve gas attacks and/or the overuse of insect repellants and pesticides such as N, N-diethyl-3-methyl-benzamide (DEET) and permethrin have been proposed. Epigenetic alterations were detected in a rat GWI model treated with pyridostigmine bromide, DEET and permethrin and exposed to mild stress, including altered methylation throughout the brain and altered expression of miRNAs and piRNAs (45). In particular, two piRNAs were upregulated in the hippocampus. The study of the GWI model implicates the involvement of non-coding RNAs and epigenetic alterations in the brain symptoms of GWI.
Associations of small non-coding RNAs with schizophrenia have also been analyzed in post-mortem samples of the anterior cingulate cortex from patients with schizophrenia. Although miRNAs and snoRNAs are differentially expressed between patients and controls, significant changes in piRNA expression were not detected in this study (46).
The findings of the aforementioned studies are summarized in Table 2, and the three most recent piRNA studies in subjects with ALS, AD and PD will be described below.
| Year | Diseases | Materials | Functions of piRNAs | References |
| 2018 | ALS | FUS (Caz) knockdown Drosophila | Pre-piRNA production is negatively regulated by FUS. Dysregulated pre-piRNAs induce cytosolic RNA-FUS aggregation along with Aub. | (15) |
| 2018 | PD | Fibroblasts, iPSCs and differentiated neuronal cells from patients with sporadic PD | SINE- and LINE-derived piRNAs are highly downregulated in fibroblasts, iPSCs and neuronal cells derived from patients with PD. | (17) |
| 2016 | GWI | Rat model of GWI in the brain | Two piRNAs (piR-007899 and piR-019162) are differentially expressed in plasma exosomes. | (45) |
| 2015 | Autism | Newborn cortical neurons from PIWIL1-inactivated rats and mice | PIWIL1 inactivation in newborn cortical neurons results in a failure of the multipolar–bipolar transition and a delay in the neuronal transition. | (41) |
| 2012 | Rett syndrome | Cerebellum in Mecp2 knockout mice | The expression of piRNAs is elevated in the Mecp2 knockout mouse cerebellum. | (40) |
| 2018 | AD | Drosophila and human postmortem brain samples | Reduced levels of piwi and piRNAs drive TE activation in a fly model of tauopathy. | (16) |
| 2017 | Prefrontal cortical tissues from patients with AD | One hundred three piRNAs were differentially expressed in patients with AD and correlates with genome-wide risk SNPs associated with AD. | (59) | |
| 2017 | AD-affected brain | One thousand nine hundred twenty-three mRNAs were significantly downregulated in subjects with AD and were the predicted targets of 125 upregulated piRNAs. | (58) | |
| 2018 | Axon regeneration after injury | C. elegans | PIWI inhibits axon regeneration. | (39) |
| 2018 | Rat sciatic nerve axoplasm | The expression of 32/53 piRNA-like RNAs is altered in the regenerating nerves. MIWI inactivation increases axon growth rates, decreases axon retraction after injury, and increases axon regrowth after injury in cultured sensory neurons. | (38) | |
| 2011 | Cerebral cortex of adult rats | piRNAs change gene expression (54 up- and 51 downregulated genes; >2.5-fold). Transcription factors control stroke-responsive piRNA expression in a redundant manner. | (37) |
ALS is a progressive motor neuron disease. In degenerating neurons, the aggregation of RNA-binding proteins such as FUS and TDP-43 is observed in cell bodies (47.48.49). In normal cells, the majority of FUS and TDP-43 is physiologically localized in the nucleus and function in RNA metabolism (50.51). In Drosophila, neuron-specific knockdown of Cabeza( Caz,an ortholog of FUS), induce movement disorders accompanied by congenital defects of the neuromuscular junctions in motor neurons, suggesting that ALS pathology is partially recapitulated (52). In this fly model of ALS, long non-cording RNAs genetically and physically interact with Caz (53). Moreover, Caz genetically interacts with piRNA-associated genes, Aub, Ago3, Wde and Rhino (15). The Caz protein binds to transcribed pre-piRNAs and suppresses pre-piRNA expression in the CNS (15). Neuronal overexpression of Aub along withCaz knockdown causes morphological defects in neuromuscular junctions and locomotor deficits in Drosophila larvae (15). Increased levels of aberrant pre-piRNAs induced by Cazinactivation accumulate in the cytoplasm with the Caz protein, which is mediated by overexpressed Aub (15) (Figure 2). Because mRNA promotes FUS multimerization (54), abnormal pre-piRNAs might stimulate Caz aggregation with Aub in the cytoplasm, which may contribute to ALS pathogenesis.
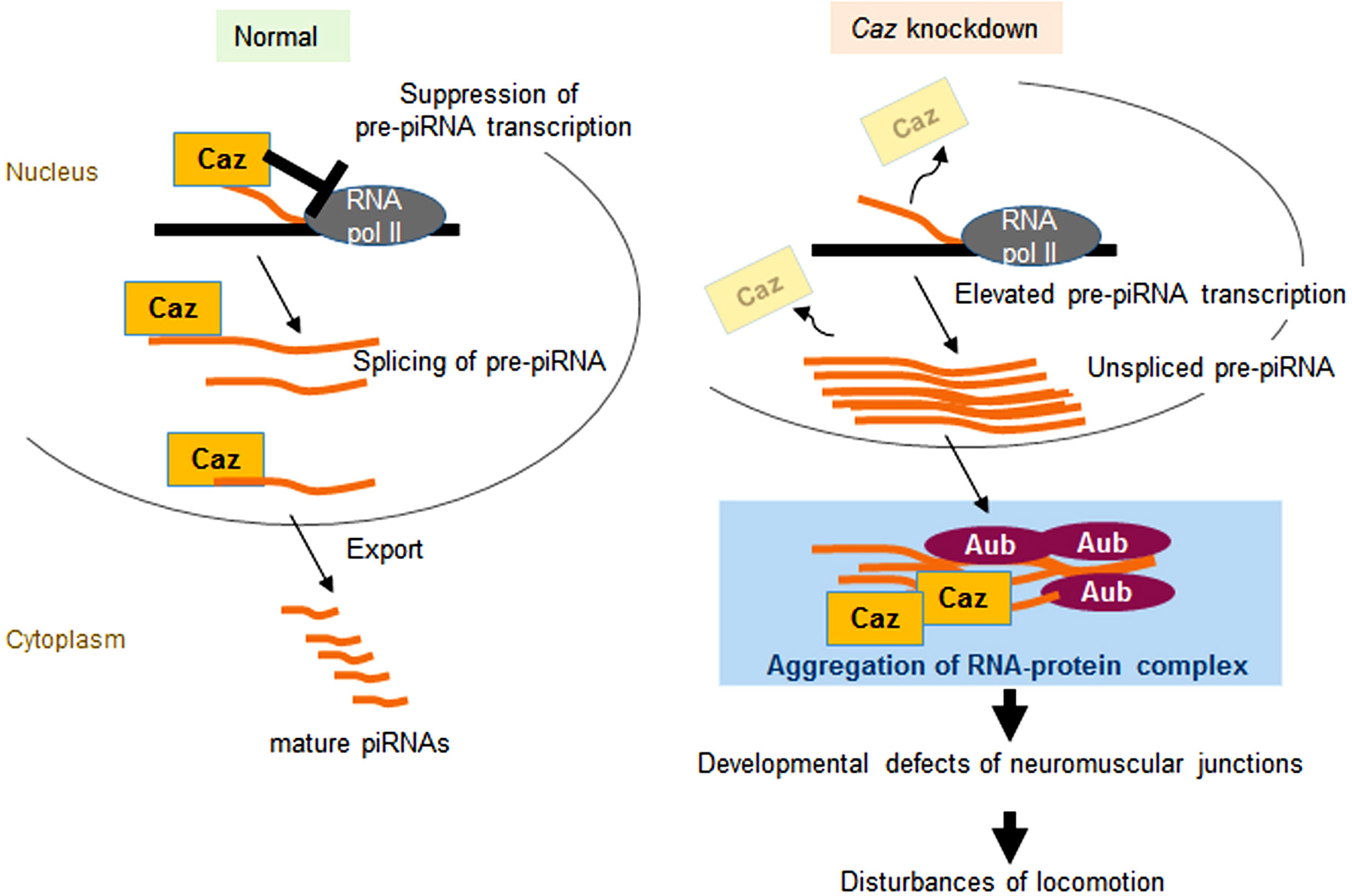 Figure 2.
Figure 2.
RNA-protein aggregation induced by aberrant pre-piRNAs in the Cazknockdown (ALS model) fly. (Left panel) In normal flies, Caz negatively regulates pre-piRNA expression and splices pre-piRNA transcripts in the nuclei. Processed pre-piRNAs are then exported by Caz to the cytoplasm to produce mature piRNAs. (Right panel) Caz suppression causes overproduction of pre-piRNAs, leading to the aggregation of RNA-protein complexes containing the remaining Caz protein in the cytoplasm, a process requiring the involvement of Aub (15).
Since abnormal pre-piRNAs and PIWI proteins have been proposed to induce the formation of RNA-protein aggregates, including FUS and TDP-43, thorough analyses of piRNA pathways in motor neurons from patients with ALS are warranted.
AD is the most common cause of dementia and is characterized by symptoms such as deficits in memory, learning and behavioral and social skills. Tau mutations are linked to AD, and neuronal Tau inclusions are thought to be a direct cause of neuron death in the hippocampus. Tau aggregation induces the decondensation of heterochromatin and depletion of Piwi in subjects with AD (55.16), allowing the activation of TEs in patients with AD (Figure 3). The combination of somatic pre-piRNA-encoding flamenco mutations and pathogenic TauR406W expression leads to aberrant cell cycle activation and neuronal cell death in Drosophila (16). Moreover, TEs are differentially expressed in the postmortem brain tissues from patients with AD and tauopathy-associated progressive supranuclear palsy. Additionally, dietary restriction and the administration of lamivudine, an inhibitor of reverse transcriptase, suppress the dysregulation of TEs and neurotoxicity induced by pathogenic tau expression in Drosophila (16).
 Figure 3.
Figure 3.
Pathogenic Tau-mediated dysregulation of the Piwi pathway in subjects with AD. Tau aggregation induces piRNA depletion and chromatin decondensation, which results in the ectopic transposition of TEs, leading to neuronal death through the ectopic activation of the cell cycle (16).
TE activation has also been implicated in motor neuron degeneration in subjects with ALS (56.57). Associations of piRNAs with AD have also reported in recent two studies (58.59). Further studies are needed to address the mechanism by which Tau aggregation induces chromatin and piRNA changes, as well as their importance in AD pathogenesis.
PD is the second most common ageing-associated neurodegenerative disorder and affects motor and non-motor neurons. The loss of dopaminergic neurons, which underlies the motor symptoms in patients with PD, is believed to be caused by mitochondrial dysfunction and/or the aggregation of the presynaptic protein /-synuclein.
The expression of piRNAs derived from short interspersed nuclear elements (SINEs) and LINEs is significantly downregulated in fibroblasts, induced pluripotent stem cells (iPSCs) and differentiated neuronal cells from patients with PD (17). The group of upregulated piRNAs in neurons differentiated from iPSCs and brain tissues was characterized by an increase in the cytosine content within the first 2-9 bp in patients with PD, although the pathological significance is unclear. Different levels of dysregulation of piRNAs have been observed in different cell types and differentiation stages (17). CpG methylation also differs among cell types, although differences have not been observed between patients and controls (17).
piRNAs have possible roles in methylating the promoter region of the CREB2 gene in Aplysia neurons, which modulates synaptic plasticity during memory formation (34). Downregulation of CREB pathway genes has also been detected in patients with PD, implying a possible connection between piRNA pathways and CREB inactivation by epigenetic modifications.
piRNA expression and DNA methylation are dynamically regulated during neuronal differentiation (17) and are involved in neuronal maturation (41). Thus, piRNAs might play important roles in various neuronal diseases through epigenetic modification of genomic DNA because the activity of TEs, which are integrated in 45% of human genomes, including the promoter regions of many genes, is regulated by piRNAs. piRNAs also represent potential therapeutic targets and appropriate diagnostic markers of neuronal diseases. Indeed, TE suppression is a potential therapeutic strategy for AD (16). Moreover, abnormally expressed piRNAs might contribute to protein aggregation in various neuronal diseases, as shown in the fly model of ALS (15). While studies on the roles of piRNAs in neuronal diseases have just begun, further investigations of piRNAs will profoundly improve our understanding of not only the biological mechanisms underlying neuronal differentiation but also the pathogenic mechanisms induced by chromatin modification.
This study was supported by a Grant-in-Aid for Scientific Research (17H04049 to Y.I.) from JSPS in Japan and was partially supported by a grant from Otsuka Pharmaceutical (Y.I.).
Abbreviations: AD: Alzheimer’s disease; ALS: amyotrophic lateral sclerosis; Aub: Aubergine; Caz: Cabeza; CNS: central nervous system; DEET: N, N-diethyl-3-methyl-benzamide; GWI: Gulf War Illness; iPSC: induced pluripotent stem cell; LINE: long interspersed nuclear element; MeCP2: methyl-CpG binding protein 2; nt: nucleotides; PD: Parkinson’s disease; pre-piRNAs: precursor of PIWI-interacting RNAs; SINE: short interspersed nuclear element; TEs: transposable elements