
Macrophages are essential elements in the tumor microenvironment, where they can promote tumor growth but also influence the efficacy of anticancer strategies. In conventional therapies, chemotherapy and radiotherapy, TAMs play a dichotomous role, contributing to antitumor activity or hindering the efficacy of cytoreductive therapies. Macrophages express checkpoint ligands and are therefore targets of immunotherapy approaches based on checkpoint inhibitors. Targeted therapies with monoclonal antibodies elicit TAMs to engage in antitumor functions such as antibody-dependent phagocytosis through the activation of Fc receptors. New approaches to exploit macrophage effector functions induced by therapeutic antibodies are under investigation. Finally, strategies aimed at targeting TAM recruitment, survival and functional polarization are advancing towards the clinic. Collectively, TAM-centered strategies will hopefully complement conventional and unconventional anticancer therapies to achieve improved therapeutic benefit.
The most frequently found cells within the tumor microenvironment are tumor-associated macrophages (TAMs) (1, 2). Due to their distinctive plasticity, macrophages are able to profoundly reprogram their functions in response to a wide variety of signals, both in physiological and pathological conditions. The combination of these signals determines the differentiation and the activation status of these cells.
The ability of macrophages to respond to different environmental cues resulting in the acquisition of distinct functional phenotypes has brought about a conventional/widely-accepted classification, referred to as M1/M2 dichotomy (3). Early studies indicated two main functional states: an activated phenotype (M1) and an alternatively activated phenotype (M2) (4). The classical M1 activation is induced by recognition of pathogen-associated moieties, such as lipopolysaccharides (LPS), via TLR ligands, and Th1 cytokines, such as Interferon gamma (INFγ), and is characterized by high levels of proinflammatory cytokines (IL1β, TNFα, IL-6, IL-23) and chemokines (CXCL9, CXCL10), increased production of reactive oxygen/nitrogen intermediates (ROI/RNI), promotion of Th1 type immune responses, and strong microbicidal and tumoricidal activity (3). Conversely, M2 macrophages respond to Th2 cytokines, IL-4 and IL-13, and are characterized by avid phagocytic activity and increased expression of scavenger receptors including CD163, mannose receptor (MR), and galactose receptor (GR), production of ornithine and polyamines via the arginase pathway, and reduced expression of inflammatory cytokines. Alternatively activated macrophages have an immunoregulatory role, and are involved in the containment of parasite (helminth) infections, tissue remodeling and tumor promotion.
While this classification might be suitable for extreme activation states such as M1 in Th1 type responses or M2 in parasite infections, and despite the fact that M1 and M2 macrophages have been detected in several pathological contexts such as sepsis, obesity, and tumors, the scenario in vivo is likely to be much more complex (4). Recently, it has been suggested that macrophages exposed to excessive stimuli, both M1 or M2, undergo a “switching” phenotype resulting in the counteractive production of mediators capable to dampen the initial stimulus (5, 6). According to this scenario, they would respond to persistent proinflammatory stimuli by reprogramming towards M2 and vice versa, integrating the type of stimulus and the phenotypic response through various intracellular signaling pathways including the JNK, PI3K/Akt, Notch, JAK/STAT, TGF-β/SMAD/non-SMAD, TLR/NF-κB (5).
In general, considering the plasticity of these cells and the complex in vivo milieu of cytokines and stimuli, often macrophages in vivo present with a mixture of these two phenotypes. It is also possible for macrophages to shift from one activation state to another during the course of an immune response or pathological context (4). Given all the above, M1 and M2 macrophages should be considered as the extremes of a single contiguous spectrum of activation states that characterize differentiated macrophages (4). Additionally, transcriptional profiling, within the “Immunological Genome Project”, of murine tissue macrophages in homeostatic conditions, found very high transcriptional diversity and minimal overlap between macrophages from different organs, strongly suggesting a highly heterogeneous and organ specific population of cells (7).
In the tumor context, studies have demonstrated that macrophages activated by bacterial products and cytokines acquire the capacity to kill tumor cells (8, 9). More frequently, though, TAMs are renowned for their promotion of tumor growth and metastasis, exerted by sustaining angiogenesis, matrix remodeling, and secreting growth factors and immunosuppressive cytokines (10–12). In accordance with these tumor-promoting functions, in many human cancer types (breast, bladder, prostate, head and neck, glioma, melanoma, and non-Hodgkin lymphoma), high numbers of TAMs have been associated to poor prognosis in preclinical and clinical studies (2, 13). However, there are some cancer types (for example colorectal and gastric cancer) in which high infiltration of TAMs correlates with better prognosis (14–16).
The wealth of studies addressing the function of macrophages and their prognostic role in human cancer sets the foundations to explore whether these important effectors of the immune response have a role in mediating the efficacy of anticancer strategies. Here we review the current understanding of how macrophages contribute to the efficacy of anticancer strategies, and, in particular, of their emerging role in targeted therapies.
Many studies have demonstrated that TAMs, besides playing a key role in tumorigenesis, also have the ability to modulate the efficacy of conventional anticancer therapies, such as chemotherapy and irradiation. Both positive and negative interactions have been documented, reflecting the complexity of the microenvironment and of macrophage functions.
The modulation of chemotherapy efficacy by macrophages is complex (Figure 1). Various mechanisms have been proposed for macrophage-mediated enhanced chemosensitivity. A first mechanism involves a process known as “immunogenic cell death” (ICD). ICD implicates the release of “eat-me” signals (for example extracellular ATP, heat shock proteins, secreted type I interferon, extracellular nucleic acids and many others still being unearthed) from tumor cells killed by cytotoxic agents, such as Doxorubicin, Oxaliplatin, Cyclophosphamide (17). These signals activate the phagocytic and antigen presenting abilities of innate immune cells, such as macrophages and DCs, which in turn are able to promote T cell responses against tumor antigens (18–22). This reinstatement of an anti-tumor immune response triggered by conventional anticancer therapies acquires particular relevance in the perspective of combinatorial strategies. Therefore, efforts are ongoing to identify anticancer agents driving bona fide immunogenic cell death to be tested in combination with immunotherapeutic strategies. Concomitantly, the attention is focused on those features of the tumor immune microenvironment that could provide an indication regarding the efficacy of the anticancer strategy, for instance the presence and density of antigen-presenting cells and their functional state.
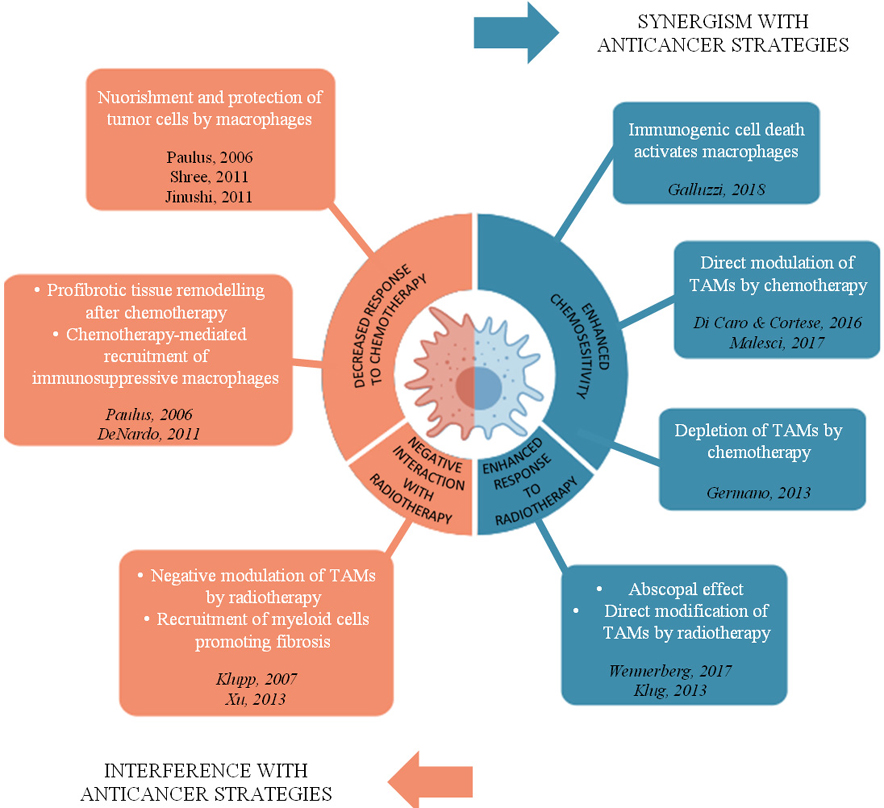 Figure 1.
Figure 1.
Distinct mechanisms mediating the interaction of macrophages with chemotherapy and radiotherapy. Macrophages contribute to the efficacy of conventional anticancer strategies by either synergizing or interfering with chemotherapy and radiotherapy. Clockwise: macrophages enhance sensitivity to selective chemotherapeutic agents inducing “immunogenic cell death”; some agents directly modulate macrophage polarization state, or selectively deplete TAMs; low-dose irradiation can drive macrophage functions to an antitumor mode, resulting in systemic tumor regression (abscopal effect) or directly modify macrophage activation. On the other hand, radiotherapy can negatively polarize macrophages or recruit pro-fibrotic myeloid cells. Interference of macrophages with chemotherapy entails macrophage capability to secrete factors nourishing and protecting tumor cells from chemotherapy, involvement of macrophages in the fibrotic reaction after chemotherapy induced tissue damage and recruitment of immunosuppressive myeloid population by chemotherapy.
A second mechanism is related to the potential effect of chemotherapy on macrophage phenotype. Both in pancreatic (15) and colorectal cancer (16), a high density of macrophages associated to a better prognosis only in chemotherapy-treated patients. The enhanced chemosensitivity hinged on the ability of the cytotoxic agents, gemcitabine and 5-fluorouracil, to reprogram macrophages from a protumoral phenotype to anti tumoral; such reprogramming, which was reflected in a change of transcriptional profile and surface marker expression towards M1, resulted in enhanced killing of tumor cells (15, 16).
Other mechanisms through which chemotherapy interacts with myeloid cells to the result of increased efficacy involve the depletion of immunosuppressive TAMs, as is the case of Trabectedin treatment for soft tissue sarcomas (23). This antitumor DNA-binding agent is selectively cytotoxic for TAMs and their circulating precursors (monocytes) by a mechanism involving selective activation of caspase-dependent apoptosis in cells of the monocyte lineage expressing the TRAIL-receptor. This macrophage-depleting effect has been shown to account for most of its antitumor activity (23). Another antitumor drug, Docetaxel, has been shown to have chemoimmunomodulating properties in a preclinical model of breast cancer (24). Here, Docetaxel treatment significantly reduced the expression of M2 markers (e.g. IL-10 and mannose receptor) and raised the expression of M1 markers (e.g. IL-12 and CCR7) in myeloid derived suppressor cells (including cells of the monocyte-macrophage lineage).
Negative effects of macrophages on responsiveness to chemotherapy are also well documented, mainly in preclinical models. The mechanisms outlined for TAM-mediated chemoresistance are often direct consequence of the most peculiar features of macrophages, namely orchestrating an immunosuppressive response, tissue-repair related functions and nourishment of tumor cells. In line with this, depletion of TAMs via anti-CSF1 antibodies resulted in enhanced chemosensitivity in a combinatorial chemotherapeutic approach in human breast cancer xenografts (25). Multiple mechanisms mediated the enhanced chemosensitivity induced by depletion of TAMs, including suppression of genes involved in chemoresistance of tumor cells and downregulation of metalloproteases contributing to tumor matrix remodeling. This evidence strongly shows the potent contribution of macrophages to tumor growth and how this can be detrimental in chemotherapeutic regimens. In a similar manner, macrophage depletion in a murine model of breast cancer was found to increase responsiveness to paclitaxel (PTX) treatment. The damage induced by PTX treatment, in fact, increased the recruitment of immunosuppressive myeloid cells, hampering the adaptive antitumor immune response (26). Other mechanisms of TAMs fostering chemoresistance involve the release of factors, such as Cathepsin B, that protect cancer cells from chemotherapy-related cytotoxicity (27), or the release of survival signals for cancer stem cells, such as milk fat globule-epidermal growth factor 8 protein (MFG-E8), limiting the effect of cisplatin on colon and lung cancer cells (28). Moreover, targeting the pro-angiogenic features of TAMs, decreasing the expression of vascular endothelial growth factor (VEGF) or placental growth factor (PlGF), resulted in ameliorated delivery of chemotherapeutic agents by improving vascular leakiness (29, 30). In colorectal cancer, the role of TAMs is still controversial. In a recent paper, macrophage infiltration associated with chemoresistance of colon cancer cell lines to 5-florouracil via the production of IL6 by TAMs which, acting on the IL6R/STAT3 axis in tumor cells, inhibited the tumor suppressor miR-204-5p (31).
Important and divergent interactions between macrophages and anticancer therapies have been shown also for radiotherapy regimens (32, 33), although molecular mediators of immunogenic cell death and type of myeloid cell activation can significantly vary compared to the ones induced by chemotherapy. Besides hitting tumor cells, in fact, radiation therapy profoundly affects the composition of the tumor microenvironment, inducing important modifications that can affect the overall type of immune response. Among others, the induction of transforming growth factor beta (TGFβ) by radiation therapy (34) holds a key role in the immunosuppressive polarization of macrophages. Also, the influx of tumor-infiltrating myeloid cells, including TAMs and myeloid derived suppressor cells, after radiotherapy has been shown to drive a fibrotic reaction that can promote tumor recurrence (35); administration of a selective inhibitor of CSF-1R has been shown to block this fibrotic reaction.
However, macrophages might be involved in the systemic “abscopal” effect induced by radiotherapy, a condition that is sometimes observed in patients when tumor regression occurs at sites distant from the irradiated lesions. The systemic reprogramming of the anti-tumor immune response induced by radiotherapy is more frequently ascribed to the antigen-presenting capability of dendritic cells, while macrophages have been more implicated as a hindrance to the radiation-induced immunity (36). However, reports have shown that neoadjuvant low-dose irradiation can elicit immunostimulatory macrophage functions, by programming the differentiation of NOS1+ M1 macrophages in human pancreatic adenocarcinoma (32, 33). Collectively, macrophages have the potential to both reduce and magnify the efficacy of radiotherapy depending on the context.
Macrophage capability to mediate efficacy of immunotherapeutic strategies has been relatively neglected, likely due to the biased view of these phagocytes as mediators of tumor progression. However, given the important contribution of TAMs to tumor biology in multiple tumor types, it is expected that macrophages and their potential involvement in immunotherapy will raise growing interest.
Only a few years after the introduction in the clinical practice of immunomodulatory antibodies, including checkpoint inhibitors, macrophages have been shown to affect their efficacy in multiple ways. Immunomodulatory antibodies target membrane molecules with regulatory functions, such as the immunological checkpoint receptors CTLA-4 or PD-1, exploited by tumor cells to evade recognition from the immune system. These antibodies have been largely supposed to act by blocking the negative signal that is induced when the receptor on T cells binds with the corresponding ligand expressed by antigen-presenting or tumor cells. However, further mechanistic studies have shown that the in vivo activity of immunomodulatory antibodies encompasses interactions with members of the FcgR family. Specifically, FcR-mediated depletion of T regulatory cells by macrophages is required in the antitumor therapeutic effects of checkpoint inhibitors directed against CTLA-4 and GITR (37, 38), suggesting that the tissue localization of macrophages in the tumor microenvironment can significantly contribute to this function. Additionally, the expression of checkpoint ligands (such as PD-L1, PD-L2 and B7-1 and B7- 2) (39–41) on macrophages makes them a key component of the immunosuppressive pathways targeted by immune-checkpoint inhibitors, and could contribute to the efficacy of these treatments (2). A recent work has unexpectedly revealed the expression of PD-1 on tumor-associated macrophages and its negative correlation with the phagocytic activity against tumor cells, suggesting another mechanism of interaction of macrophages with checkpoint inhibitors (42). Prompted by the increasing number of studies assessing the role of macrophages in checkpoint treatments, numerous clinical trials are now testing the efficacy of therapeutic regimens combining blockade of macrophages by CSF1/CSF1-R inhibitors and of checkpoint axes (43).
Very recently, histo-pathological analysis of non-small cell lung cancer specimens from patients treated with anti-PD1 in neoadjuvant regimen has evidenced macrophages in the regression bed (the area of immune-mediated tumor clearance) and their assessment has been used to build an irPRC (immune-related pathologic response criteria) scoring system (44) which could be exploited to identify standardized assays to assess immunotherapeutic efficacy.
Macrophages have also been implicated in the mechanism of action of agonistic antibodies targeting the co-stimulatory receptor CD40, by a mechanism involving polarization towards an antitumor mode of action (45). The original studies proposing macrophages as target cells of the CD40 agonists opened a new vista on the potential exploitation of these phagocytes for therapeutic purposes. In the wealth of efforts aimed at depleting macrophages in cancer, Beatty and colleagues showed that, when properly re-educated by immunomodulating agents, CD40 agonists in this case, macrophages turned into a favorable element in the tumor microenvironment, suggesting that reprogramming strategies could be efficacious approaches to enhance other forms of immunotherapy. CD40 agonists are now being tested in combination with chemotherapy, checkpoint inhibitory antibodies, and other immune modulators (46).
The number of therapeutic antibodies targeting tumor-specific antigens currently being used in the clinical setting is growing. Their mode of action encompasses different mechanisms, including block of tumor survival signals, neutralization of tumor growth factors, complement activation and engagement of effector immune cells expressing the FcR (47–49).
The strategic tissue localization and the high concentration of macrophages in the tumor microenvironment (Figure 2) of many cancer types make these phagocytes ideal mediators of the efficacy of antibody-based targeted therapies. Early studies from the 1980’s assessed and demonstrated the contribution of macrophages to the mechanism of action of monoclonal antibodies in vivo (50–52); however, whether the exact mode of action was phagocytosis of tumor cells rather than cytotoxicity or a mixture of both was not very clear. As professional phagocytes, in fact, macrophages recognize and engulf harmful materials by specialized receptors, including pattern recognition receptors, scavenger receptors and receptors for the Fc (fragment crystallizable) portion of antibodies (53); therefore, they are fully equipped to perform important anti-tumor functions such as phagocytosis (54) and tumor cell lysis (49).
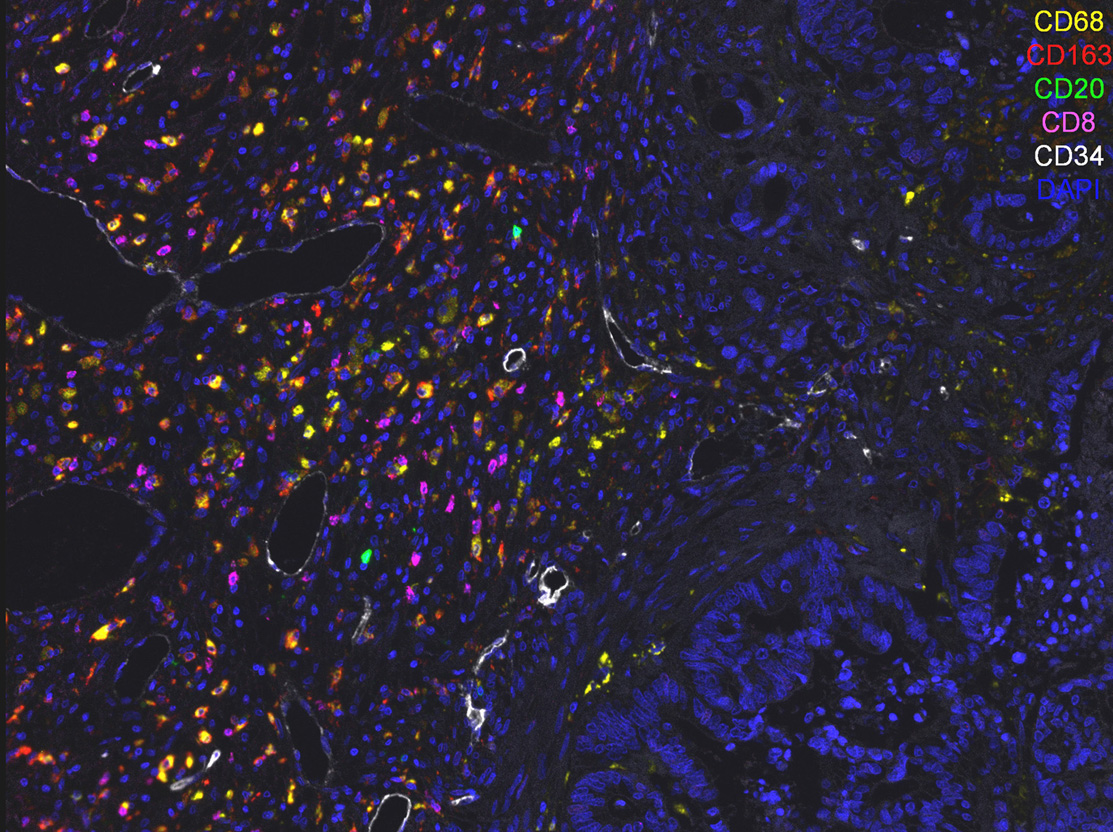 Figure 2.
Figure 2.
TAMs within the tumor microenvironment. TAMs accumulate in high numbers in the tumor microenvironment, making them ideal candidates for therapeutic approaches and important effectors of targeted therapies. CD68+ and CD163+ (yellow and red respectively) macrophages, CD20+ B cells (green), CD8+ T cells (magenta) and CD34+ vessels (white) in a human section of colo-rectal liver metastasis.
From an immune cell standpoint, the therapeutic antibody serves to connect the target cell with the activating type I FcR, primarily expressed on effector natural killer cells and phagocytes. Effector cells are activated to induce antibody-dependent phagocytosis (ADP) of the opsonized target or exert cytotoxicity through the release of cytotoxic mediators and antibody-dependent cell-mediated cytotoxicity (ADCC). Examples of therapeutic antibodies for which effector macrophages expressing the FcR are involved include Rituximab, targeting the CD20 B cell-differentiation antigen expressed by lymphoma and leukemia B cells (55, 56), Trastuzumab, an antibody against the erb-b2 receptor tyrosine kinase 2 (ERBB2 or HER2) (47, 57–59), Cetuximab, targeting the epidermal growth factor receptor (EGFR) (49), Daratumumab targeting CD38 in myeloma cells (60). Accordingly, functional polymorphisms in human FcgRIIIA have been identified that may affect the ADCC of natural killer cells and monocyte/macrophages and correlate with response rates in lymphoma patients treated with Rituximab (61), breast cancer patients treated with Trastuzumab (59) and metastatic colo-rectal cancer treated with Cetuximab (62).
An important confirmation of the relevant role of macrophage phagocytosis induced upon recognition of therapeutic antibodies comes from the studies on the CD47/SIRPα axis. SIRPα is a negative regulator of macrophage phagocytosis and its engagement by CD47, widely expressed by many cell types, is perceived as a “don’t eat me signal” by the phagocytes. Overexpression of CD47 by tumor cells (63–66) has been implicated in resistance of tumor cells to macrophage phagocytosis and its therapeutic targeting is being evaluated in clinical settings (67). Notably, the inhibition of macrophage phagocytic activity by the CD47/SIRPα axis suggests that targeting of this axis could complement antitumor antibodies and possibly reinforce their efficacy. This hypothesis has been tested and proven effective. CD47-blocking antibodies have shown synergistic activity in combination with Rituximab (63) and non-functional engineered SIRPα variants have been used as adjuvants for antitumor antibodies including Rituximab, Cetuximab and Trastuzumab (68).
Based on all the preclinical and clinical evidence highlighting a tumor-promoting role for macrophages, in recent years, many strategies targeting TAMs have been proposed. Mainly, the approaches have been focused on impeding the recruitment of monocytes/macrophages to the tumor site (by blocking the CCL2/CCR2 or the CXCL12/CXCR4 axes), interfering with differentiation and survival (targeting the CSF-1/CSF-1R pathway), and acting on polarization.
Monocyte recruitment to tumor tissues relies on several molecules including chemokines (CCL2/CCR2 and the CXCL12/CXCR4 axes), CSF-1 and VEGF. Several preclinical models of melanoma, breast, liver, lung, and prostate cancer treated with specific antibodies anti-CCL2 have shown a reduction in tumor growth and metastasization (2). In clinic however, interfering with the CCL2/CCR2 pathway has given contradictory and rather unsatisfactory results in terms of efficacy, even if generally well tolerated (69, 70). This could be due to the robustness of the chemokine family and compensatory mechanisms involving other macrophage-recruiting chemokines. CNTO88 (carlumab), a specific, inhibitory monoclonal antibody anti-CCL2, entered a phase I clinical trial for advanced solid tumors, but only showed a transient CCL2 suppression (71). Another phase I clinical trial was conducted with carlumab in combination with different chemotherapy regimens for patients with solid tumors, but again only transient depletion of CCL2 was achieved and no increased efficacy compared to chemotherapy alone (69). However, in a phase I study for pancreatic adenocarcinoma, another selective CCR2 inhibitor (PF-04136309) showed more promising results in combination with chemotherapy. Approximately 50% of patients receiving the combined treatment showed partial tumor response (70).
In a similar manner, targeting the CSF-1/CSF-1R pathway has yielded better results in combination regimens rather than alone. CSF-1 is the main cytokine for differentiation and survival for the monocyte/macrophage lineage. With the additional advantage of the receptor (CSF-1R) being expressed exclusively on monocytes, it represents an evident candidate to target macrophages. CSF-1 is also over-expressed by many tumor types, and its expression correlates with bad prognosis in various cancer types (2).
In experimental models, the monoclonal antibody RG7155 (Emactuzumab), which targets CSF-1R, was able to reduce the influx of TAMs and skew the adaptive immune infiltrate towards CD8 lymphocytes (72). Another molecule targeting CSF-1R, PLX3397 (Plerixafor) was tested for recurrent glioblastoma and demonstrated improved efficacy only when in combination with radiotherapy (73). Furthermore, in a preclinical transgenic model of pancreatic adenocarcinoma, the CSF-1R inhibitor GW2580, enhanced chemosensitivity to gemcitabine (74).
Finally, as already mentioned in the previous paragraph, macrophage targeting to the aim of functional activation was unexpectedly observed after treatment with a CD40 agonist antibody in a small group of patients treated with gemcitabine. This was reproduced in a genetically engineered mouse model of pancreatic cancer, with the result of reduced tumor growth, due to repolarization of macrophages towards a tumoricidal phenotype. Surprisingly, the mechanism was shown to be both T-cell and gemcitabine independent (45). This promising study led to a phase I clinical trial on a small group of advanced pancreatic cancer patients, testing the CD40 agonist antibody (CP-870,893) in combination with gemcitabine, which yielded only a partial tumor response (75).
A more recent study has shown that targeting the Bruton Tyrosine Kinase (BTK) pathway in a preclinical model of pancreatic cancer can lead to reactivation of the adaptive immune response, via repolarization of TAMs towards an M1-like phenotype (76).
Taken together, the preclinical and clinical evidence convincingly indicate that targeting TAMs will bring best results when in combination with standard therapies.
Macrophages are essential elements of the immune ecosystem of tumors, often present in high numbers and characterized by a peculiar malleability, which allows them to acquire both protumor and antitumor functional states. Most studies have documented the relevance of the macrophage population as a whole; however, dissecting the heterogeneity of TAMs could provide new insights on their role in the tumor microenvironment.
TAMs are important mediators of the efficacy of anticancer strategies (Figure 3). In conventional therapies, chemotherapy and radiotherapy, macrophages have been shown to both boost and limit the therapeutic effect. These observations suggest that strategies aimed at reprogramming macrophages towards an anti-tumor phenotype could yield promising results in combination with conventional cancer therapies. However, preclinical and clinical evidence varies significantly in a context-dependent manner, occasionally highlighting contradictory roles for TAMs.
 Figure 3.
Figure 3.
TAMs are important mediators of the efficacy of anticancer strategies. Synergism between TAMs and conventional therapies, chemotherapy and radiotherapy, encompasses mechanisms such as imunogenic cell death, TAM depletion and TAM reprogramming. Interference of TAMs with therapeutic effect includes orchestration of tissue fibrosis and M2-like functions. The in vivo activity of immunomodulatory antibodies encompasses also interactions with members of the FcgR family, expressed on macrophages. Macrophages express checkpoint ligands (such as PD-L1, PD-L2), thus representing a key component of the immunosuppressive pathways targeted by immune-checkpoint inhibitors. In targeted therapies, monoclonal antibodies engage macrophage antitumor functions, such as their ability of eliminating cancer cells by antibody-dependent phagocytosis and cytotoxicity.
Recent studies have evidenced a role for TAMs in immunomodulatory and targeted anticancer therapies. Monoclonal antibodies engage macrophage antitumor functions, such as their ability of eliminating cancer cells by antibody-dependent phagocytosis and cytotoxicity. Therapeutic strategies exploiting the engagement of macrophages to unleash the full potential of the innate immune system are now approaching the clinic. Hopefully, such strategies will complement conventional therapies to reach improved therapeutic benefits.
The research leading to these results has received funding from Associazione Italiana per la ricerca sul cancro (AIRC) under IG2016-ID.18443 project – P.I. Marchesi Federica and AIRC fellowship 18011 to Nina Cortese. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. Authors declare no conflict of interest. The Authors declare no competing interests.