
Academic Editor: Baohong Zhang
Ribonucleic acid (RNA) can act as a hapten in the direct immunization of animals. For antigen synthesis, 65 mg of viroid RNA were obtained by in vitro transcription of the recombinant DNA. We received a reasonable immune response in mice and rabbits with synthesized conjugate viroid RNA-lysozyme. Analyses of polyclonal mouse and rabbit antisera as well as estimates of antibody specificity were performed by dot-Enzyme Linked Immunosorbent Assay (ELISA), sandwich ELISA, and northern immunoblotting. Antiserum obtained showed strong cross-reactions with cellular RNA. The viroid polyclonal antibody cross-reactions with cellular RNAs were depleted via titration antibodies by the plant cellular or commercial yeast RNA. We successfully used antibodies against the viroid RNA-lysozyme antigen to detect the wild-type potato viroid and diagnose potato viroid infection. We presume that intrinsic cross-reactions of RNA antibodies are potentially dangerous after nucleic acid vaccination. Research into the specificity of antibodies against viral RNAs is underway.
Interest in antibodies specific to nucleic acids has a long history. Activity in this area was immense because antibodies to DNA and RNA were detected in the serum of patients with viral infection or diagnosed with multiple autoimmune diseases [1, 2, 3, 4, 5, 6, 7]. In the case of direct immunization of animals with nucleic acids, it became evident that natural nucleic acids act as haptens [1]. Nevertheless, antibodies to Z-DNA [8], RNA [3, 4, 5, 7], synthetic and natural double-stranded RNA [9, 10], modified nucleotides in RNA [11, 12, 13, 14, 15], and cellular RNA [16, 17] were obtained.
There are few reports of antibodies’ production against natural RNA [10, 16, 17, 18].
A specially selected recombinant mAb against brain cytoplasmic 200RNA (BC200)
with a Kd of
In extended RNAs, antibodies recognize primary [3, 4, 15, 16, 17, 18], secondary [5, 9, 10, 11], and tertiary structures [18]. Hu et al. [19] published a comprehensive review of antibodies to synthetic and natural nucleic acids and their applications. Those antibodies specifically recognize up to six nucleotides of the RNA primary structure; the immunogenicity of dsRNA is higher than that of ssRNA, and antibodies can recognize the nucleic acid helixes’ structure.
The interaction of antibodies with the RNA antigen involved formation of hydrogen bonds between polar amino acid residues in the hypervariable regions of the antibodies; phosphate, oxygen, and hydroxyl groups of the furanose ring; as well as exocyclic amino, and carbonyl groups of the nucleotides in the RNA. The formation of antigen-antibody complexes might also engage stacking interactions of aromatic rings of amino acids (Tyr, Trp) and nucleotide bases.
We studied the specificity of polyclonal antisera directed against RNA-VPg and
the synthetic covalent linkage unit (CLU) Tyr-(5
Among the infectious nucleic acids, the most intriguing is viroid RNA (vdRNA) [23]. The lack of a protein capsid has long been the reason for the impossibility of using serological methods for the viroids detection. To date, the most common methods for the molecular diagnostics of plant virus infections are immunological. The diagnostic approaches for detecting viroid infections are based on molecular hybridization of nucleic acids mainly using DNA probes and polymerase chain reaction (PCR) [24]. Lukacs et al. [16] reported monoclonal mouse antibodies toward the potato viroid. They cross-reacted 25 synthetic and natural nucleic acids, and the highest affinity was for ribosomal RNA.
Despite prior unsuccessful attempts, the goal of this work was to obtain polyclonal antibodies that recognize vdRNA specifically. Notably, the immune response was expected to result in polyclonal antibodies against several ribotopes of the RNA antigen. Polyclonal antibodies’ palette of recognized ribotopes of the viroid complex antigen will be broader than any monoclonal antibody; therefore, polyclonal antibodies have to display higher avidity. In this regard, it was essential to study the cross-reactions of polyclonal antibodies to viroid RNA with host cellular RNAs because the similarity of RNA molecules at all levels of structural organization is much closer than that of the protein (see Discussion).
Deionized double distilled water (TDW) was used throughout this work; stock
buffer solutions were filtered through a 0.22-
Healthy and PSTVd-infected potatoes were kindly provided by Dr. N.V. Girsova (All-Russian Research Institute of Phytopathology, Moscow, Russian Federation). An Apple Scar Skin Viroid (ASSVd) preparation was presented by Dr. V. Hallan (CSIR-Institute of Himalayan Bioresource Technology, Palampur, India). The identity and purity of the PSTVd-transcript, ASSVd, RNA, RNA-antigens, and antibodies preparations obtained in this work were confirmed by the PSTVd cDNA sequencing, UV-Vis spectroscopy, gel-electrophoresis, dot-ELISA, sandwich ELISA, and immune northern blot assay. UV absorption spectra were recorded in the range 220–320 nm using Thermo Scientific NanoDrop 2000 Spectrophotometer (Thermo Fisher Scientific).
Here, 1 g of fresh or frozen S. tuberosum leaf tissue was thoroughly
ground with a mortar and pestle in liquid nitrogen. The leaf powder was extracted
with 6 mL of buffer solution (1% SDS, 100 mM NaCl, 10 mM EDTA-Na
The cDNA copy of the Russian potato viroid isolate [26] was obtained using the
following primers: (+)PSTVd 5
We previously labeled RNA with (dien)platinum for multiplex determination of
viruses and PSTVd in infected potatoes with nucleic acid probes [28]. To obtain a
vdRNA-(dien)platinum complex, the PSTVd-transcript was denatured for 3 min at 95
Borate buffer (248
Agarose gel electrophoresis was performed in 1.5% agarose gels using a buffer
composed of 40 mM Tris-acetate, 2 mM EDTA (pH 7.5), and 0.5
Laboratory outbred mice and female Chinchilla rabbits were immunized. Pre-immune
sera were used as a control in the ELISA screening after immunization and stored
at –70
The globulin fraction of the antiserum was obtained by fractionation with ammonium sulfate [31]. Immunoglobulins against the vdRNA-lyso antigen were isolated from the globulin fraction by affinity chromatography on the Hi-Trap Protein G column (GE Healthcare, Boston, MA, USA) according to the manufacturer.
The concentration of horseradish peroxidase was calculated according to
A
Antigens and RNA samples were adsorbed and cross-linked to 0.22
MaxiSorp Nunc-Immuno plates (Swedesboro, NJ, USA) were coated with the capture
primary polyclonal antibodies against antigens (1
Antigens and RNA samples in water were electrophoresed in 2% agarose gels in
TAE buffer with ethidium bromide (0.5
It is challenging to extract sufficient viroid from infected potatoes. PSTVd
accumulates in the infected potato to less than 1
 Fig. 1.
Fig. 1. Characteristics of the PSTVd-transcript, its antigens, and Apple
Scar Skin Viroid (ASSVd). (A) UV-spectra; colored curves: black—baseline.
Green—PSTVd-transcript: A
(Dien)platinum reacts with RNA [28, 34]. We previously modified the viroid RNAs with (dien)platinum under conditions where predominantly guanine bases were modified [29] for diagnostics of virus infections [28]. We considered the chemical modification of the PSTVd-transcript with both (dien)platinum and lysozyme to be stochastic. Regarding the antigens, we assumed that modification of the PSTVd-transcript with (dien)platinum causes minimal structure distortion because the melting temperature of the modified RNA probes, UV-spectrum, and electrophoretic mobility in the agarose gel (Fig. 1B) changed only insignificantly.
(Dien)platinum also reacts with DNA, and this modification enhanced its antigenic properties [29]. Moreover, we assumed that, along with antibodies to the adduct of (dien)platinum and guanine residues, part of antibodies would also recognize nucleotides adjacent to this adduct. However, the titer of mice antiserum against the vdRNA-(dien)Pt antigen was significantly lower than that against the vdRNA-lyso antigen (Fig. 2); therefore, we stopped working with this antigen.
 Fig. 2.
Fig. 2.Comparison of mice sera and antisera against antigens vdRNA-(dien)Pt and vdRNA-lyso. Top line: (A) Antiserum after seven immunizations with vdRNA-(dien)Pt antigen. (B) Antiserum after 3 immunizations with vdRNA-lyso antigen. From left to right, quantities of antigens applied to the membranes: (1) 100 ng; (2) 20 ng; (3) 4 ng. Bottom line: (A,B)—mice pre-immune sera.
The choice of lysozyme as a booster protein for the synthesis of the RNA-antigen was made earlier [22]. This basic protein (pI 9.3) readily forms complexes with RNA and partially protects RNA from nucleases. Linking with lysozyme produced more visible changes; most products of the reaction smeared close to the gel load wells far from the position of the vdRNA (Fig. 1C). There are both fast and slow kinetics of the formaldehyde reaction with nucleic acid bases [35] and proteins [36, 37, 38, 39] above neutral pH. Depending on the reaction conditions, reactions with formaldehyde occur in a few minutes whereas the slow reactions take from hours to days [35, 39]. We assumed that subsequent protein and nucleic acid modification reactions change their conformation bringing together (and moving aside) the functional groups that form the methylene cross-links between the protein and the nucleic acid.
Three main parameters play a critical role during formaldehyde cross-linking:
reaction temperature, incubation time, and formaldehyde concentration. Room
temperature is advantageous because it presents the most pertinent condition.
Considering the ionization constants of the amino acids [40] and nucleic
base functional groups, the conjugation reaction between lysozyme and vdRNA was
performed at pH 9.3. In this mild alkali medium, the RNA is stable, and all
heterocyclic bases readily interact with formaldehyde. The reaction proceeds
through the formation of methylol derivatives and reactive Schiff bases [35]. We
assumed that formaldehyde could potentially react with several amino acid
residues of the egg white lysozyme [41]: The first is the primary amino group of
N-terminal lysine and the
The Russian isolate of PSTVd used in the present work consists of 357 nucleotides (71A, 78 U, 108 C, and 100 G) and has a molecular mass of ~114,733 [26]. We selected stoichiometric binding conditions of (dien)platinum and lysozyme to RNA in both cases considering reacting chemical groups, and we used reaction conditions that minimize profound RNA modification (Fig. 1B,C). Under mild antigen-preparation conditions, part of the transcript remained unconjugated to lysozyme. Therefore, the concentration of the vdRNA-lyso antigen preparation was determined by the content of its main component, the transcript.
Electrophoresis in 1.5% agarose: (B) (1) the vdRNA-(dien)Pt antigen. GeneRuler, 1kb DNA Ladder (Thermo Fisher Scientific); (2) the PSTVd-transcript; (3) the vdRNA-(dien)Pt antigen; (C) (1) the vdRNA-lyso antigen. RiboRuler low range RNA ladder (Thermo Fisher Sci., bases); (2) the PSTVd-transcript modified with lysozyme; (3) the PSTVd-transcript. The upper band is the viroid dimer. (D) (1) ASSVd, 7% polyacrylamide gel electrophoresis. Trace amount of the PSTVd-transcript was added; (2) DNA ladder (base bairs, Thermo Fisher Scientific GeneRuler 1kb Plus); (3) the PSTVd-transcript.
We found that the vdRNA-lyso antigen was much more immunogenic than the vdRNA-(dien)Pt antigen (Fig. 2). The immune response was weak even after seven immunizations of mice with the vdRNA-(dien)Pt antigen (Fig. 2A). A more robust response was observed after the third immunization of mice with the vdRNA-lyso antigen (Fig. 2B). Therefore, we immunized a rabbit with this antigen only.
The results indicated that immunization with RNA preparations is more challenging than immunization with protein antigens. According to our protocol, at least eleven injections were required to obtain a viroid antibody titer sufficient for reliable detection. A comparison of the immune responses suggests that lysozyme act as an internal booster of the immune system that significantly accelerates the immune response to the antigen’s RNA component.
Dr. Lukacs reported that monoclonal antibodies against PSTVd cross-react with
cellular RNA—particularly ribosomal RNA [16]. Notably, in an infected cell, the
mass of cellular rRNA is hundreds of times superior to the mass of PSTVd. The
dot-ELISA assay of specificity of antiserum and purified antibodies against the
vdRNA-lyso antigen (Figs. 3C,4) showed that both cross-react with lysozyme and
potato cellular RNA. In the polyclonal antiserum, complete blocking of antibodies
to lysozyme was achieved by adding 1.5 mg/mL lysozyme. To completely block
cross-reacting cellular antibodies, we added commercially available yeast RNA or
cellular potato RNA to the polyclonal antiserum or the antibodies (Fig. 3). The
minimal required amount of the yeast and cellular RNA was determined
experimentally. As a result, a linear diagnostical signal intensity dependence in
the range from 1
 Fig. 3.
Fig. 3.Dot-ELISA of the rabbit antiserum to the vdRNA-lyso antigen
after suppressing the antiserum cross-reaction by lysozyme (A and B) and yeast
RNA (C). Membranes with samples were incubated in the immune (upper line) and
pre-immune (bottom line) sera with added 1.5 mg/mL lysozyme: (A) the
vdRNA-lyso antigen; (B) PSTVd-transcript. Both A and B membranes were spotted
with 1.0
 Fig. 4.
Fig. 4.Recognition of the PSTVd-transcript by antibodies to vdRNA-lyso
antigen. Lysozyme (1.5 mg/mL) and the cellular yeast RNA (0.15 mg/mL) were added
to the polyclonal antibody preparation to suppress cross-reactions with lysozyme
and cellular RNAs. Samples from left to right: 1.0, 0.3, and 0.1
We hypothesized that antibodies to the PSTVd-lyso antigen recognize not only nucleotide(s) covalently associated with lysozyme but (oligo)nucleotides adjacent to the points of modification. The results in Fig. 4 confirm this supposition.
To detect PSTVd in the infected potato, we removed high-molecular-weight cellular rRNAs from the cleared lysate of the viroid-infected plant cells (see Methods) by 2 M LiCl precipitation. We then concentrated the low-molecular-weight RNA through precipitation from the LiCl supernatant using ethanol. Dot-ELISA of the antiserum to the vdRNA-lyso antigen confirmed that it contains antibodies interacting with lysozyme, the antigen, and the unmodified PSTVd-transcript (Figs. 3,4). The immunogenic reactivity towards lysozyme was neutralized by adding 1.5 mg/mL lysozyme. We confirmed that the primary source of cross-reactions is ribosomal RNAs [16]. The cross-reaction with the plant’s cellular RNAs was suppressed by saturation cross-reactive antibodies with 0.15 mg/mL yeast RNA. We found that the sensitivity of detecting the viroid RNA-transcript (up to 60 ng/mL, Fig. 5) was sufficient to determine natural viroids in a sample of low-molecular-weight RNA from infected potatoes. The data above are supported by quantitative methods of the sandwich-ELISA with the isolated antibodies against the vdRNA-lyso antigen (Fig. 5).
 Fig. 5.
Fig. 5.Sandwich ELISA of the RNA preparations with pre-immune serum and isolated antibodies against the vdRNA-lyso antigen. Pre-immune serum. Colored curves: Brown (dotted line)—low-molecular-weight RNA of the uninfected potato. Black (triangular blank markers)—low-molecular-weight potato RNA with the addition of the free PSTVd-transcript (10 ng/mL). Violet (square blank markers)—PSTVd-transcript.
Isolated antibodies. Blue (round black markers)—the low-molecular-weight RNA of the uninfected potato; Green (triangular black markers)—the low-molecular-weight RNA of uninfected potatoes with the addition of the free PSTVd-transcript; Yellow (square black markers)—low-molecular-weight RNA from the PSTVd-infected potatoes; and Red (solid line without markers)—the free PSTVd-transcript.
The dependence of the signal intensity of the immunochemical reaction on the concentration of vdRNA in the low-molecular-weight fraction of the total RNA of infected potatoes (yellow line in Fig. 5) is linear from 30 to 250 ng/mL with a sample standard deviation of 0.02–0.04. Thus, the results suggest that vdRNA is immunogenic; however, the antibody-specific titer is moderate partially because the RNA might undergo intense hydrolysis in physiological fluids.
Diagnosis of viroid infections has largely been solved with the invention of the polymerase chain reaction. However, the PCR reaction requires sample preparation. Sample preparation, PCR, and RT-PCR require laboratory equipment and trained personnel. In agriculture, PCR technology is used only in specialized laboratories in highly developed countries. Historically, most of the mass diagnostic tests in practice are carried out by immunochemical methods in agriculture. Moreover, current trends in the development of molecular diagnostics of infections are focused on bringing these methods closer to their use by non-professionals or ordinary manufacturers of products; thus, they are used directly in the field for culling infected plants or animals. The use of immune chromatography test strips facilitates this. Even in the field, immune chromatography methods can diagnose pandemic, epizootic, and epiphytotic diseases [42]. This method’s simplicity, speed, and scalability make it practical and convenient for analyzing many samples.
The specificity of antibodies against RNA was determined by comparing them with antibodies to proteins. Proteins consist of 20 different amino acids, and thus there are very many combinations. There are also many ways that an antibody can recognize an epitope on a protein. It does not need to be a consecutive sequence of amino acids, i.e., it can be part of two or more patches of the three-dimensional architecture of the amino acid sequences—this is done simply by forming the recognized part. Polyclonal antibodies can react with several but slightly differing epitopes. A similar is observed with ribotopes.
While the building blocks of RNA are limited to four bases, post-transcriptional modifications can dramatically extend the chemical diversity of RNA with up to 170 identified modifications [13, 15]. Moreover, the tertiary structure of RNA is also specifically recognized by anti-RNA antibodies along with the primary and secondary structures [18]. Although antibody binding sites span a 7–10 linear nucleotide sequence, their avidity was found to increase for elongating nucleotide sequence [16, 17, 19]. In single-stranded DNA, the antibody’s avidity for a hexanucleotide increases up to 300 nucleotides [17, 19].
Nevertheless, the antibodies’ specificity for nucleic acids is lower than that of antibodies to proteins—the polyclonal antibodies to RNA manifest cross-reactions with other nucleic acids. The RNA’s ordinary primary structure degeneration versus the protein’s primary structure explains the bulk of cross-reactions of these antibodies with other RNAs. The target ribotopes can be identified using blocking (subtracting) cross-reacting ribotopes by appropriate RNA. This access is conceptually similar to subtractive hybridization. Thus, neutralized polyclonal antibodies against the PSTVd-lyso antigen that cross-reacts with cellular RNA can detect PSTVd RNA in infected cells (Figs. 3,4,5,6). Specific immunoglobulins using ribotope subtraction can selectively recognize a closely related apple viroid (Fig. 7) and can be discovered among those raised against the PSTVd-lyso antigen.
 Fig. 6.
Fig. 6.Electrophoretic and immune northern blot analyses of the
PSTVd-transcript and the PSTVd-infected potato. Left—electrophoresis of RNA
samples in a 1.5% agarose gel in the presence of ethidium bromide. Left row:
numbers show the length of the marker RNAs in nucleotides. (1) Marker RNAs of the
Thermo Fisher Scientific RiboRuler Low Range RNA Ladder; (2) low molecular RNA
isolated from healthy potato (10
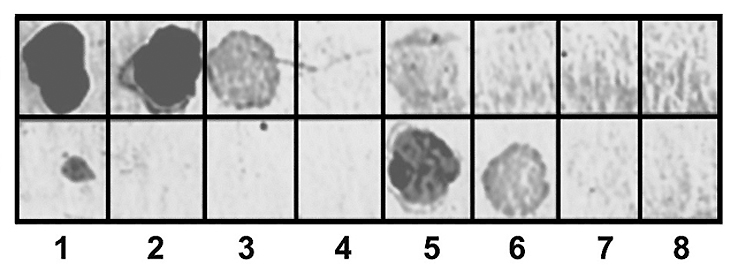 Fig. 7.
Fig. 7.Dot-ELISA of the specificity of antibodies against the
PSTVd-lyso antigen. Similar quantities: Upper line—1, 0.3, 0.1, and
0.03
The immune response against even simple viroid RNA is pleiotropic due to this macromolecule’s plurality of structural ribotopes. The advantage of polyclonal antibodies obtained in this work over monoclonal antibodies (used by Dr. Lukacs) is their higher overall avidity towards the antigen due to binding to all ribotopes at once. Notably, immunoglobulins recognizing the target RNA, despite intrinsic cross-reactions, can be used in immune analysis.
The specificity of the antibodies was investigated through northern immunoblotting. Fig. 6 shows that the antibodies react specifically with the PSTVd-transcript obtained in vitro and the potato spindle tuber viroid in the low-molecular-weight RNA preparation of infected potato.
We currently obtained a population of polyclonal immunoglobulins with different
specificities, and each recognizes its ribotope. The antibodies were raised
against stable RNA structures. Notably, even small viroid RNA is a large
macromolecule (
For comparative analysis, the same quantitative dilutions of the PSTVd-transcript and apple viroid were applied to the membrane (Fig. 7).
Fig. 7 shows that the antibodies against PSTVd-lyso interacted differently with the PSTVd-transcript and ASSVd. They recognized the PSTVd-transcript but not the apple viroid. Moreover, the polyclonal antibody preparation appears to contain a fraction of immunoglobulins that cannot be saturated with the PSTVd-transcript, but these recognize ASSVd. Thus, despite their close relationship, these viroids appear to have similar and different ribotopes recognized by different immunoglobulins in the preparation of polyclonal antibodies against the PSTVd-lyso antigen.
Both viroids (PSTVd and ASSVd) belong to the Pospiviroidae family and have similar secondary structures (Fig. 8, Ref. [23, 43]).
 Fig. 8.
Fig. 8.Primary and secondary structures of PSTVd and ASSVd. Sequentially, the viroid domains for PSTVd and ASSVd are presented according to references [23] and [43]. T1, left terminal domain; P, pathogenicity domain; C, central domain; V, variable domain; and T2, right terminal domain. Vertical dashes show domain boundaries.
The apple scar skin viroid (ASSVd) consists of 329 nucleotides. It differs strikingly from all potato spindle tuber viroid (PSTVd)-related viroids in that its central domain shows no sequence similarity with the central conserved region of the latter [43]. Thus, there is different recognition of PSTVd and ASSVd (see Fig. 7); this is mainly explained by the differences in their primary structure. We assume that the specificity of antibodies to the target RNA can be significantly increased if, instead of RNA, one uses the ribotopes with the specific primary structure as RNA antigens. Thus, it would be interesting to know the differences in the ribotopes of viral and cellular RNAs.
Furthermore, we observed that the resulting polyclonal antibodies have cross-reactions with cellular RNAs—mainly with rRNA. This polyclonal antibody property is critical to the recently increased interest in vaccine mRNAs. The developers’ primary attention is on the vaccine properties of viral proteins encoded by the vector mRNA. Studies that assessed the immunogenicity of messenger RNA and vector DNA vaccines showed that the vaccines were safe with no evidence of short-term allograft-related adverse effects in either cohort.
One might wonder if an mRNA (DNA) vaccine would lead to an overactive immune response. Humans have innate immunogenicity against foreign RNA [44, 45] and DNA [1, 2, 7], but the specificity of antibodies for nucleic acids is much lower than that of antibodies for proteins. RNAs are usually weaker antigens than proteins but not their complexes with proteins. RNAs form complexes with proteins in cells. In this regard, the likelihood of undesirable formation of antibodies to RNA increases sharply. This is undesirable due to the more remarkable similarity of the RNA composition of the pathogen and the host versus proteins.
We speculate that antibodies to mRNA and DNA can cross-interact with cellular RNAs and host DNA because of their chemically close PAMPs (Pathogen-Associated Molecular Pattern). Furthermore, foreign high molecular weight mRNAs are exposed to nucleases and the RNA interference system during vaccination; they are then degraded as many RNA complexes in the cell. Degradation occurs to those mRNAs in which natural nucleotides have been partially replaced with nuclease-resistant analogs and even those bound to polyamines, lipids, dextran, and placed into liposomes. The resulting mRNA fragments that lost their ability to translate are still immunogenic.
Our preliminary data with the plant and animal virus RNAs support this supposition. Thus, this is a likely pathway to mRNA and vector DNA vaccination’s unfortunate consequences—an autoimmune disease [44, 45]. The cause of the disease is that antibodies produced in response to bacterial or viral nucleic acid during infection (vaccination) begin to bind to host nucleic acids, their regulatory sequences, and even proteins due to molecular mimicry [46, 47]. In most cases, this leads to a disruption of the cellular mechanisms that control our bodies. Autoimmune diseases develop slowly but are relatively common. We suspect that the current practice of vaccination using mRNA and DNA vaccines as well as inactivated and live viruses have not had sufficient time (in the case of mRNA) to study the long-term consequences of nucleic acid vaccination. The long-term side effect of the nucleic acid vaccination requires additional study in our opinion [48].
Finally, diverse functional RNAs participate in a wide range of cellular processes. The RNA structure is critical for function—both for RNA on its own and as a complex with proteins and other ligands. Therefore, analysis of RNA conformation in cells is essential for understanding their functional mechanisms. However, no appropriate method has yet been established, and there are few practical tools for recognizing the conformation of structured RNA in vivo. Antibodies against RNA can fill this gap, thus helping to recognize specific RNA conformations. To be closer to this purpose, the definition and identification of the PSTVd and some virus RNA’ ribotopes are underway. Furthermore, antibodies against target ribotopes can potentially be a magic bullet that regulates in vivo RNA activity.
ABTS, 2,2
YD, TG, and KB conceived and designed the experiments; TG and KB performed the experiments; TG and KB contributed reagents and materials; YD and KB analyzed the data.
All experiments on animals (mice, rabbits) were carried out in accordance with the animal care regulations of the Moscow State University by Lomonosov. The protocol was approved by the Bioethics Committee of the Faculty of Biology, Moscow State University by Lomonosov.
We thank D. Blaas and A.G. Gabibov for critically reading the manuscript draft. Valuable tips and comments of E.V. Skurat and E.S. Gavryushina are appreciated. Furthermore, we thank our undergraduate students A. Koroleva and E. Goldobina, for their technical assistance. The authors sincerely thank N.V. Girsova, T.B. Kastalyeva, and K.A. Mozhaeva (Institute of Phytopathology, Moscow region) for valuable advice and providing the tubers of the healthy and infected with viroid potatoes. The authors are grateful to V. Hallan for the preparation of apple viroid. This research has been supported by the Interdisciplinary Scientific and Educational School of Moscow University ”Molecular Technologies of the Living Systems and Synthetic Biology”.
This work was supported by the grant of the Russian Science Foundation No. 16-16-04108.
The authors declare no conflict of interest.