
The plant proteins called ERECTA family play important role in inflorescence
architecture, stomatal patterning and phloem-xylem organization. ERECTA proteins
belong to the moonlighting proteins family containing the guanylyl cyclase (GC)
catalytic center embedded within the intracellular kinase domain. This
characteristic architecture of ERECTA proteins prompted us to experimentally
confirm of enzymatic activity of one of these, BdERL1 (ERECTA-like1 from
Brachypodium distachyon). We have shown that BdERL1 is dual-function
protein with both kinase and GC activity. Moreover, our mutagenesis studies also
revealed the catalytic roles of key conserved amino acid residues at the GC
center and importantly, probing of the kinase and GC with Ca
The ERECTA family of genes, including ERECTA (ER) and ERECTA-LIKE1,2 (ERL1,2), encode leucine-rich repeat receptor-like kinases (LRR-RLKs) in plants. The most studied ERECTA gene family from Arabidopsis thaliana plays important roles in many physiological processes. ERECTA kinases are involved in stomatal development, the regulation of longitudinal growth of aboveground organs, reproductive development and plant responses to biotic and abiotic environmental stress factors [1]. The interaction between the ERECTA family and plant hormone signaling (such as auxin, gibberellins or ethylene) has also been confirmed [2, 3, 4, 5]. Interestingly, a few years ago, three receptor kinases belonging to the ERECTA family from A. thaliana, At2g26330 ERECTA (ER), At5g62230 ERECTA-like1 (ERL1), and At5g07180 (ERL2), were bioinformatically selected as potential guanylyl cyclases (GCs) [6]. In silico identification of a 14 amino acid (aa) long catalytic center, [KS]X[SCG]X(10)[KR]X(0,3)[DHSE], within these proteins suggests that they are able to convert GTP into cGMP. However, biochemical proof of the GC activity of these kinases still needs to be discovered. The amino acid sequence of ERECTA proteins indicates that they belong to the moonlighting family of proteins containing a GC catalytic center embedded within the intracellular kinase domain. Many of these moonlighting proteins are receptor kinases that show a cytosolic dominant function, kinase functionality, an additional cryptic function, cyclase activity, and more importantly, regulatory roles between and within the proteins localized to their cellular microenvironments, which are poorly understood [7, 8]. As such, many of the biological processes and responses affected by this class of second messengers have not been fully elucidated.
In this report, we describe the biochemical characterization of BdERL1 from Brachypodium distachyon (NCBI accession no. XP_003561134.3). The purpose of our study was to establish the moonlighting function of the GC in BdERL1 and to determine the catalytic roles of the two functionally assigned and highly conserved amino acids at positions 3 and 14 within the GC catalytic center of BdERL1, as well as to investigate how a functional GC in BdERL1 might affect its kinase activity.
The studied fragment of BdERL1 cDNA (NCBI no. XM_003561086.4) was
amplified by PCR using the following primers:
5
The PCR product was introduced into the pGEX-6P-1 expression vector linearized
with BamHI and NotI restriction enzymes using In-Fusion cloning
technology (In-Fusion HD Cloning Kit, Clontech). The correctness of the cloned
cDNA sequence was verified by sequencing. The E. coli BL21 strain,
transformed with the resulting plasmids, was used to produce GST-tagged proteins.
Cells were grown in LB medium supplemented with 2% glucose at 37
The homogeneity and purity of the protein fractions were analyzed by 10% (v/v) SDS–PAGE, and the gels were stained with Coomassie blue.
It was previously described that functionally assigned residues of the GC motif
can be replaced with another amino acid to estimate the importance or relevance
of these residues to maintain the functional configuration of the catalytic
center [7]. The G residue localized at position 3 within the GC catalytic motif
has been predicted to determine substrate specificity for GTP. We mutated the G
residue (842) in the catalytic center to D (mutant G
Site-directed mutagenesis was performed using a QuikChange II
XL site-directed mutagenesis kit (Agilent). A nonmethylated
double strand was synthesized using 125 ng of forward
(5’-gagcaactctagaagaacgat
The guanylyl cyclase activity of the studied proteins was determined by
estimating the rate of cGMP formation. For the BdERL1 WT and mutant assay, the
reaction mixture contained 50 mM Tris-HCl buffer (pH 7.5), 5 mM MgCl
Analogically, the cross reactivity of the studied proteins with ATP as a substrate at concentrations between 0.25–2 mM was analyzed.
A negative control with the pure GST protein was performed to exclude its cyclase activity.
Liquid chromatography–tandem mass spectrometry (LC-MS/MS) was used as the method for the GC/AC activity assay. The enzymatic activity was defined as the amount of cGMP/cAMP produced by 1 mg of protein per minute.
LC-MS/MS experiments were performed using the Nexera UHPLC and LCMS-8045
integrated system (Shimadzu Corporation). The ionization source parameters were
optimized in positive ESI mode using pure cGMP or cAMP dissolved in HPLC-grade
water (Sigma). Samples were separated using a reversed-phase C18 column (150
The calculation of cyclic nucleotide content was based on standard curves for cGMP and cAMP ranging from 0 to 200 pg (0–0.7 pmol).
For quantification of the BdERL1 protein kinase activity, a Kinase-Glo plus
luminescent kinase assay (V3771, Promega) was used according to the
manufacturer’s protocol. Briefly, purified intracellular domain of BdERL1 (1
Domain predictions were performed using InterPro
(https://www.ebi.ac.uk/interpro/) and LRRfinder
(http://www.lrrfinder.com/lrrfinder.php). The Met
A highly conserved and stringent GC search motif for higher plants
[KS]X[SCG]X(10)[KR]X(0,3)[DHSE] was constructed, and its continuous refinement in
recent years has allowed for the discovery of a number of functional candidate
GCs in the plant kingdom [13, 14, 15, 16, 17, 18, 19, 20]. Based on bioinformatics prediction of the
GCPred tool (http://gcpred.com), the BdERL1 protein was
identified as a potential new guanylyl cyclase harboring a conserved GC motif
that is localized within the cytosolic C-terminus of the studied protein (BdERL1
GC center
 Fig. 1.
Fig. 1.Structural features of GC catalytic domain of BdERL1. (A) Representation of domain organization of BdERL1 containing a leucine-rich repeats (LLRs), transmembrane domain (TM), and fourteen amino acid long search motif GC motif embedded in kinase domain. (B) Amino acid sequence of BdERL1 fragment showing the kinase domain (grey shaded) with GC center (underlined and red) and potential CaM binding domain (underlined and green). Arrows indicate two functional residues in the GC catalytic center at 3 and 14 positions mutated for functional studies of BdERL1.
The full-length amino acid sequence of BdERL1 belonging to the leucine-rich-repeat receptor kinase family shows very high homology to other kinases of monocotyledonous plants, such as Hordeum vulgare (92% amino acid sequence identity), Aegilops tauschii (92%), Triticum aestivum (92%), Oryza brachyantha (91%), Panicum miliaceum (91%), and Sorghum bicolor (90%). Lower similarity (~70%) was observed between BdERL1 and LRR-RLKs belonging to dicotyledonous plants such as Camellia sinensis (76%), Eucalyptus grandis (76%), Vitis vinifera (76%), Ricinuscommunis (73%) or A. thaliana (72%). However, a high level of identity among the plant ERECTA-like protein family is noticeable regardless of their divergent phylogenetic evolution.
Alignment of the BdERL1 aa sequence with the three most experimentally studied ERECTA proteins from A. thaliana (AtER At2g26330; AtERL1 At5g62230; AtERL2 At5g07180) revealed the highest (72%) identity with the AtERL2 protein. All of these proteins possess a 14 aa long GC motif with highly conserved functional residues and architecture characteristics for moonlighting kinases. The significant level of similarity suggests common (moonlighting) functions for these proteins in both plant species. It was revealed that AtERL2 is involved in the determination of epidermal cell division, i.e., whether the cell should divide proliferatively to produce pavement cells or divide asymmetrically to generate stomatal complexes [22]. The GC activity has not been studied in any ERECTA protein from A. thaliana thus far; therefore, BdERL1 is the first experimentally confirmed guanylyl cyclase belonging to the ERECTA family.
We performed computational assessments using structural and molecular docking strategies to determine the catalytic feasibility of the BdERL1 GC domain. Since the BdERL1 kinase domain has 44% amino acid identity to AtBRI1 and an even higher identity at the corresponding GC center (78%), we modeled BdERL1 against the AtBRI1 crystal structure (PDB ID: 5LPV) [9]. We show in this model that the GC catalytic center is solvent exposed, allowing for substrate interactions and also presumably for catalysis (Fig. 2A). Furthermore, the GC center of BdERL1 contains a characteristic alpha-helix barrel followed by a loop secondary fold that is an innate feature of experimentally validated plant GCs and ACs identified by this motif-based approach [20]. Further probing of the GC center by molecular docking of GTP suggested that GTP can dock at the GC center with a good free energy value (–5.6 kcal/mol) and a favorable binding pose where the negatively charged phosphate end of GTP points toward K853 (position 14 of the motif) while the guanine end is surrounded by negatively charged residues including S840 (position 1 of the motif) (Fig. 2), similar to the structurally resolved GC centers [23]. We also performed the same docking simulation with ATP and found that the mean binding affinity was lower than that obtained with GTP (data not shown).
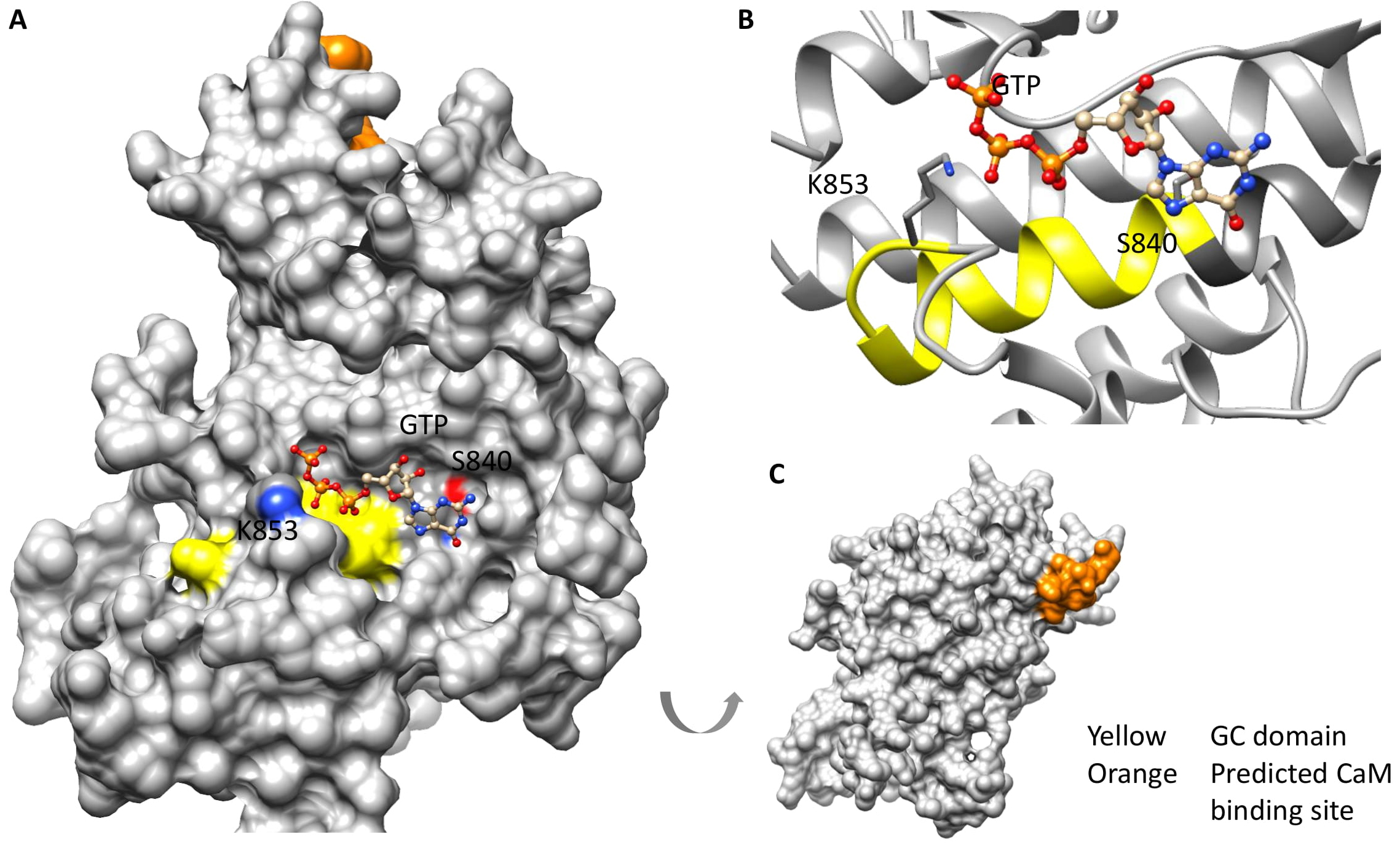 Fig. 2.
Fig. 2.Computational assessment of the BdERL1 GC domain. Docking of GTP to the GC center of BdERL1 and the interaction of GTP with the key residues at the catalytic center (yellow) is shown as (A) surface and (B) ribbon models, respectively. Conserved key amino acids at position 1 (S840) and position 14 (K853) of the motif are shown as individual residues and labelled accordingly in the models. (C) The predicted CaM binding site (orange) is located at the rear of the kinase and GC domains.
To test whether BdERL1 is able to generate cGMP in vitro, the fragment
(BdERL1
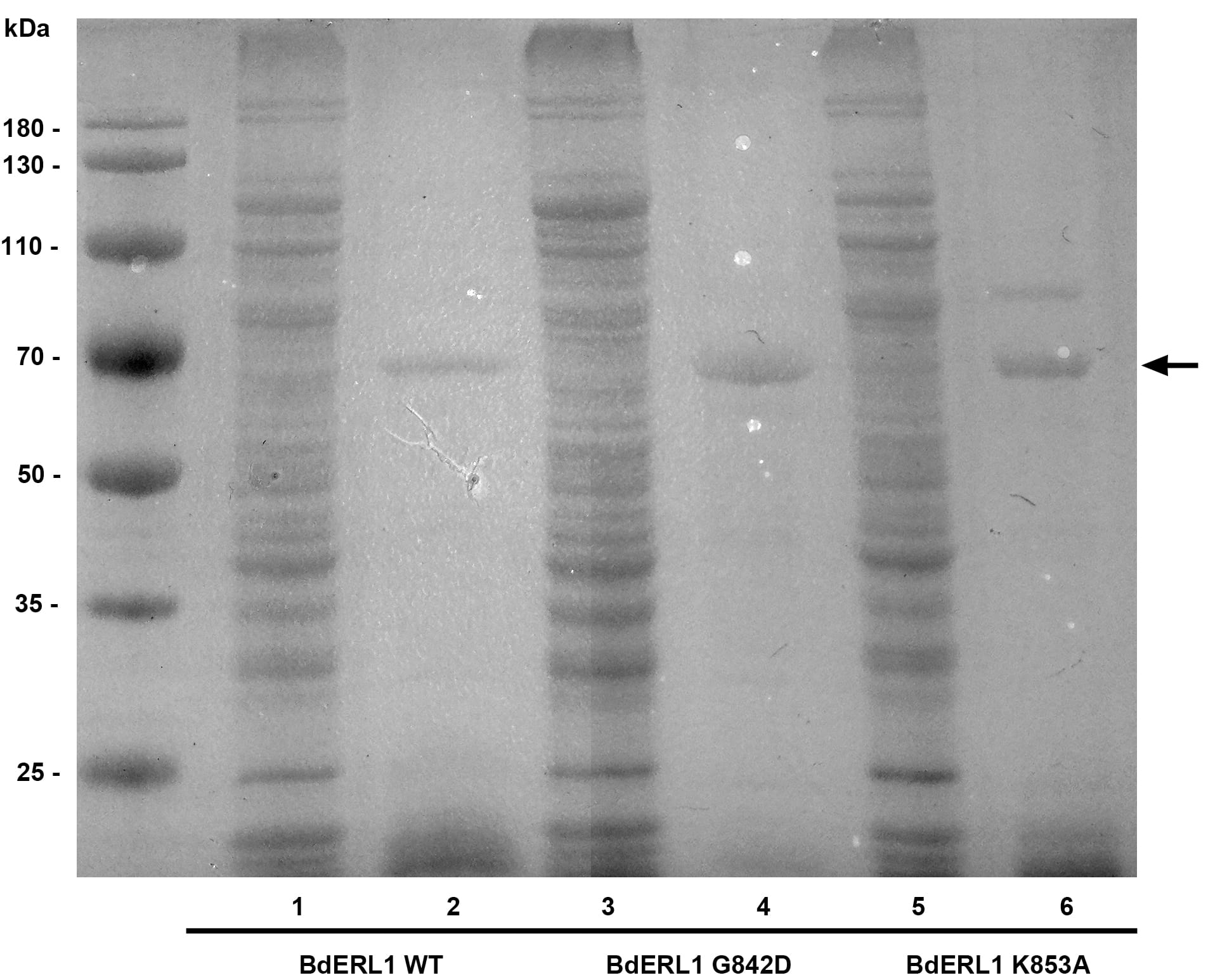 Fig. 3.
Fig. 3.SDS-PAGE of BdERL1 WT, BdERL1 G
Experimental analysis confirmed the bioinformatics prediction of the webtool
GCPred [21] as well as the subsequent computational assessments involving both
structural assessments and molecular docking simulations, thus suggesting that
BdERL1 can act as a functional guanylyl cyclase. The highest GC activity of the
BdERL1 protein was observed in the presence of 1.5 mM GTP, magnesium, and
manganese ions, where the cGMP concentration reached its maximum of
~70 pmol mg protein
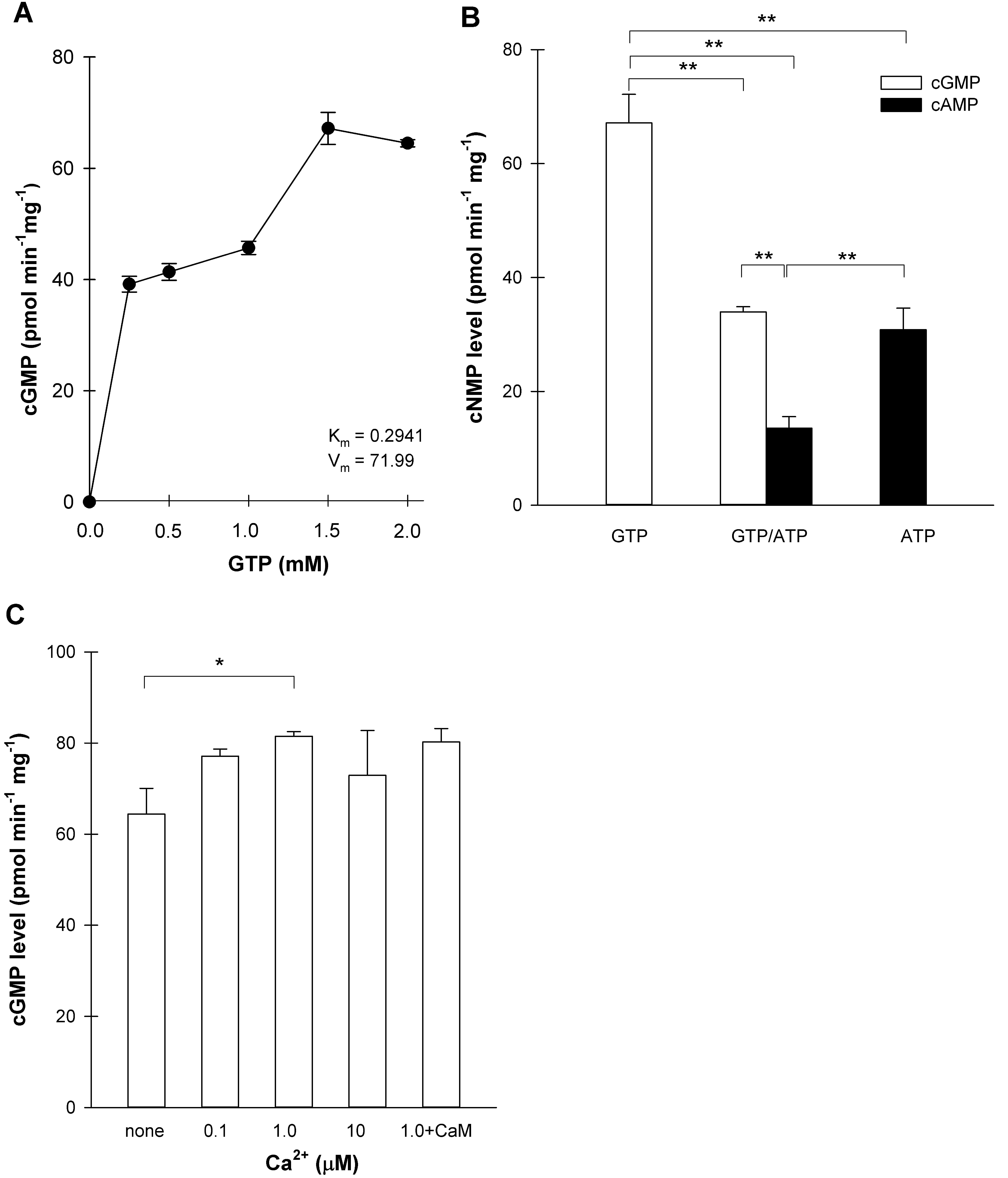 Fig. 4.
Fig. 4.Enzymatic guanylyl cyclase activity of recombinant BdERL1 WT.
(A) Recombinant BdERL1 WT activity in response to various concentrations of GTP.
Reaction mixture contained 50 mM Tris/HCl buffer (pH 7.5), 5 mM MnCl
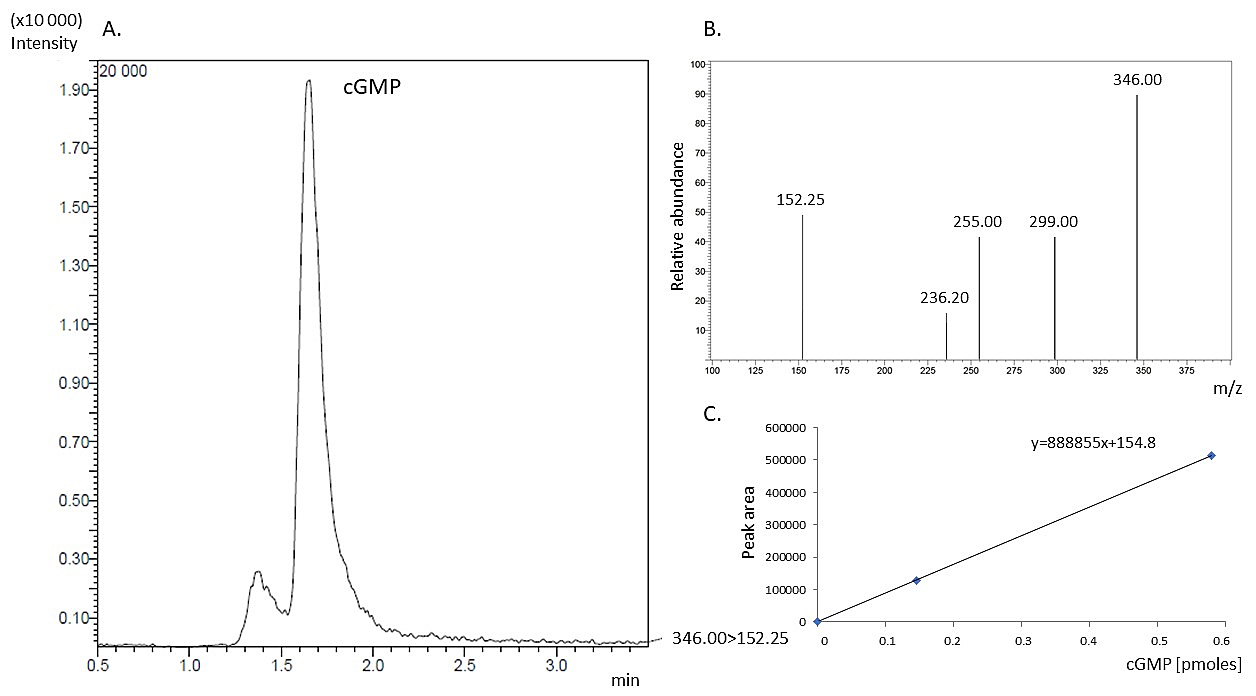 Fig. 5.
Fig. 5.Detection of cGMP generated by BdERL1 by LC-MS/MS. (A)
Determination of guanylyl cyclase activity of the BdERL1 protein by LC-MS/MS. Ion
chromatogram of cGMP was generated from a reaction mixture containing 5
Moreover, the BdERL1 protein was also able to convert ATP to cyclic AMP, but the catalytic activity was approximately two times lower than that with GTP as the substrate (Fig. 4B). This suggests a significant reduction in affinity for ATP at the catalytic center, which is consistent with the data obtained from the molecular docking simulations, which also predicted a lower binding affinity for ATP compared to GTP at the catalytic center. Cross-reactivity is not uncommon for plant GCs and ACs since substrate discrimination is normally poor for moonlighting centers, and this innate characteristic has been described in previous reports [13, 16, 17]. Notably, the addition of an equivalent amount of ATP and GTP (1:1 ratio) into the reaction mixture generated cGMP and cAMP in amounts that were both lower than those achieved by their respective substrates alone, although the concentration of the cAMP product remained two times lower than that of cGMP, thus clearly showing a preference for GTP over ATP as the substrate (Fig. 4B). Since nucleotide triphosphates share very similar size and structural features, ATP could compete with GTP for the binding site at the GC center.
Previous reports have shown that the activity of plant transmembrane GCs called
moonlight proteins can be modulated by calcium ions [28, 29, 30]. In
vitro experiments have shown that the GC activity of the phytosulfokine receptor
AtPSKR1 is significantly enhanced by calcium at physiological levels (0.1–10
The wild-type (WT) BdERL1 protein (without amino acid sequence changes) and two
BdERL1 proteins with mutations to the GC catalytic center were generated. It was
previously described in an animal retinal guanylyl cyclase that substitution of
the amino acid that confers substrate specificity can convert a GC into an AC
[32]. In the case of plant GCs, the amino acid at position 3 of the motif was
hypothesized to confer substrate specificity [20, 33], and therefore, we mutated
this amino acid (G
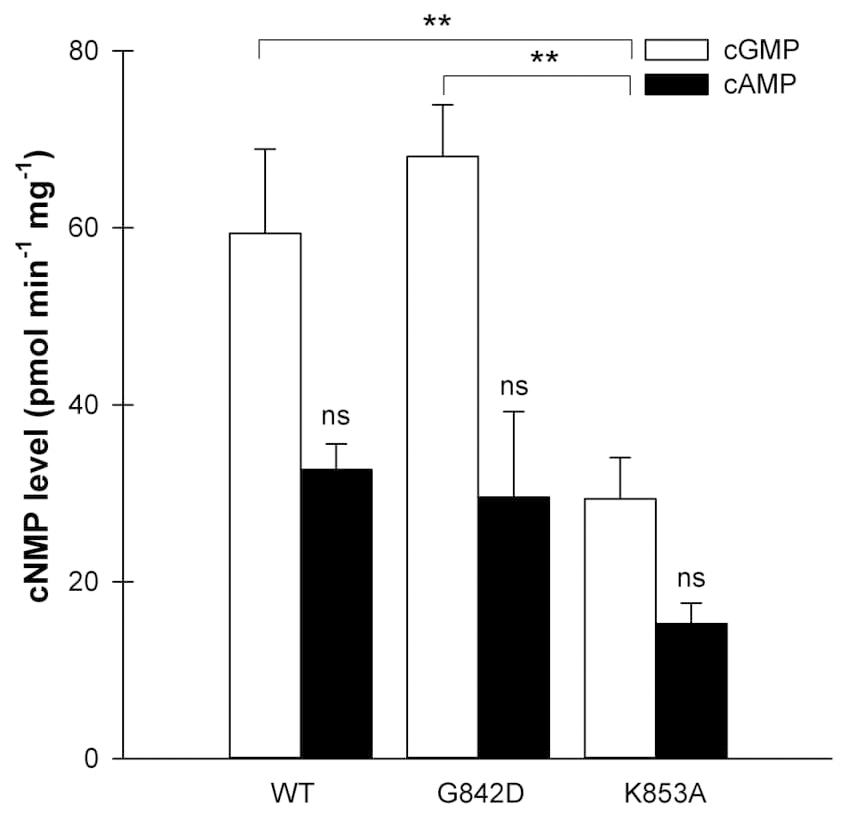 Fig. 6.
Fig. 6.Detection of cGMP and cAMP generated by BdERL1 WT, BdERL1
G
In a second mutated BdERL1 protein, the amino acid lysine (K) at position 14 was
mutated to alanine (A) (K
The architecture of previously characterized LRR-RLKs reveals that they are
moonlighting proteins with GCs embedded within the primary kinase domain [36, 37], and sequence analysis has also disclosed the same domain organization for
BdERL1. Here, we showed that BdERL1 is also a functional kinase, and more
importantly, its activity is inhibited by cGMP (Fig. 7A). The addition of cGMP at
concentrations of 0.1 or 1
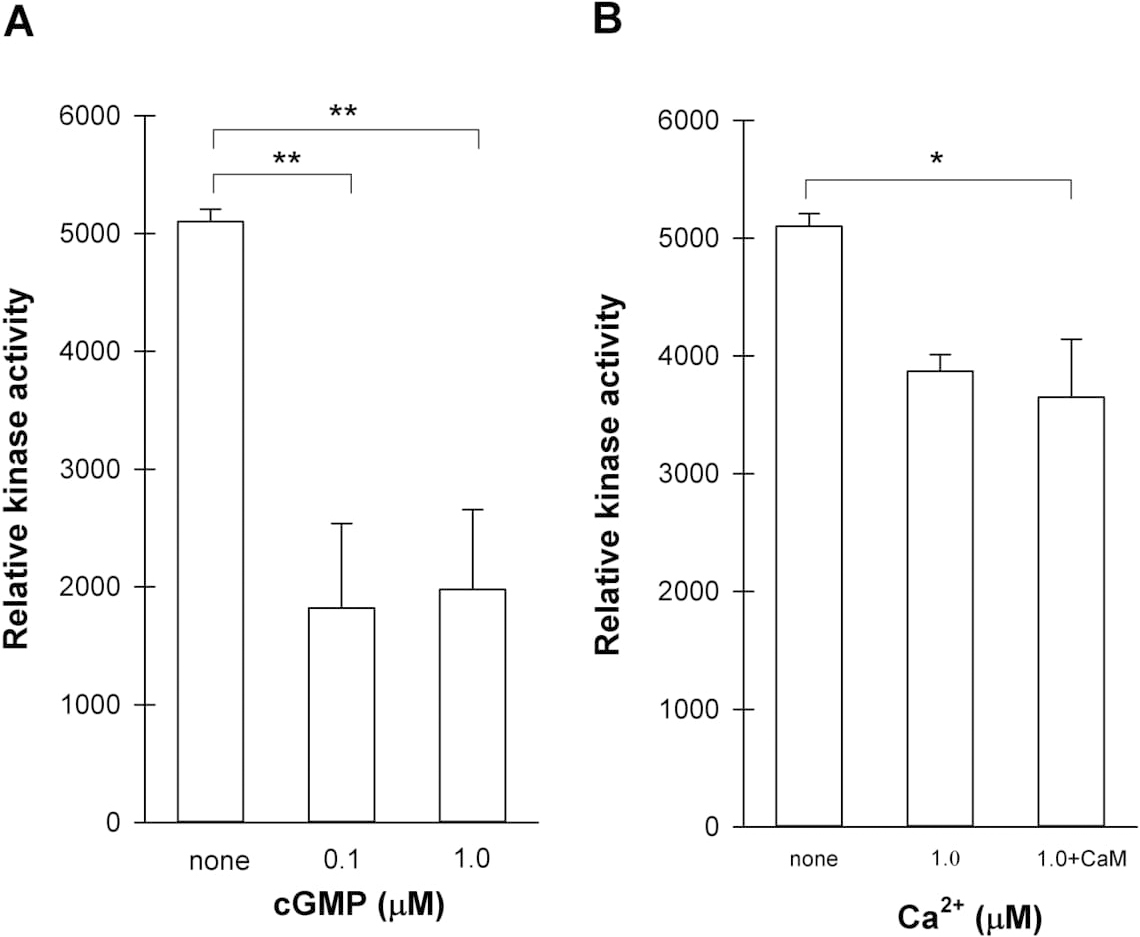 Fig. 7.
Fig. 7.Determination of kinase activity of recombinant BdERL1 WT by
Kinase–Glo plus luminescent kinase assay. (A) BdERL1 WT kinase activity in
response to various concentrations of cGMP. Reaction mixture contained 25 mM
Tris/HCl buffer (pH 7.5), 5 mM MgCl
Calcium ions were previously proposed to be a bimodal switch between GC and the
kinase activity of AtPSKR1 [29]. The kinase activity of AtPSKR1 was directly
inhibited by free calcium ions, and its guanylyl cyclase activity was enhanced at
similar calcium concentrations. Consistent with the findings of AtPSKR1, the
molecular switch function of calcium also operates in BdERL1, where Ca
In conclusion, we employed a combination of computational and experimental
approaches to identify a functional GC that is embedded within the kinase of
BdERL1. Moreover, our mutagenesis studies revealed the catalytic roles of key
conserved amino acid residues at the GC center. Importantly, probing the kinase
and GC with Ca
All authors have read and approved the final manuscript. Conceptualization—BŚB and ASJ; methodology—BŚB and MD; formal analysis—BŚB, MD, KH and AW; investigation—BŚB; writing—original draft preparation—BŚB; writing—review & editing—MD, AW, KJ and ASJ; visualization—KJ; supervision—KJ and ASJ; funding acquisition—BŚB, MD, AW.
Not applicable.
Not applicable.
This work was supported by National Science Centre (Poland) grant number 2018/29/N/NZ9/00812 PRELUDIUM15 and 2018/02/X/NZ1/00103, MINIATURA2), and funds provided by Nicolaus Copernicus University (Toruń, Poland) for the Research Program of the Chair of Plant Physiology and Biotechnology. A.W. was supported by the National Natural Science Foundation of China (32100581) and the Zhejiang Provincial Natural Science Foundation of China (LQ19C130001).
The authors declare no conflict of interest.