
Academic Editor: Paolo Ivo Cavoretto
Objectives: Hypertensive disorders occur in approximately 12% to 22% of pregnancies and cause substantial perinatal morbidity and mortality of both mother and fetus. Hypertensive disease is directly responsible for approximately 20% of maternal deaths and can be classified as chronic hypertension, gestational hypertension, preeclampsia-eclampsia, and chronic hypertension with superimposed preeclampsia. At present, the pathogenesis of preeclampsia is still unclear, we wrote this article to make a uptodate review of this disease. Mechanism: A comprehensive search of several databases was conducted from inception up to March 2022. The searched databases were Web of Science, MEDLINE,Ovid, and Cochrane Database of Systematic Reviews. The search strategy included the combinations of the following medical terms: Hypertensive disorders; preeclampsia; mechanism; pathogenesis hypothesis. Findings in Brief: At present,the pathogenesis of preeclampsia is still unclear, the theory of Genetic,Inflammatory Response, Immune Imbalance in Maternal-Fetal Interface, Oxidative Stress, Vascular Endothelial Cell Damage are supposed involved in the progress of preeclampsia. Conclusions: Although there are various theories mentioned above, none of the hypothesis can fully explain preeclampsia. More research is needed on the mechanism of preeclampsia.
Hypertensive disorders occur in approximately 12% to 22% of pregnancies and cause substantial perinatal morbidity and mortality of both mother and fetus. Hypertensive disease is directly responsible for approximately 20% of maternal deaths and can be classified as chronic hypertension, gestational hypertension, preeclampsia-eclampsia, and chronic hypertension with superimposed preeclampsia [1, 2].
Preeclampsia is the development of hypertension with proteinuria after 20 weeks of gestation, with or without proteinuria, in conjunction with fetal growth restriction (FGR), maternal endothelial dysfunction, and chronic immune activation [3]. The main risk factors for the development of preeclampsia (PE) are first pregnancy, previous or family history of PE, chronic hypertension, diabetes, antiphospholipid syndrome (APS), obesity and thrombophilia (Table 1, Ref. [4, 5]). The basic pathophysiological changes are systemic arteriole spasm, vascular endothelial injury and ischemia, causing damage to multiple organs [3].
| Risk factors | Odds ratio (OR) or Relative risk (RR) (95% CI) |
| Antiphospholipid antibody syndrome | 9.7 (4.3–21.7) |
| Renal disease | 7.8 (2.2–28.2) |
| Preeclampsia in previous pregnancy | 7.2 (5.8–8.8) |
| Systemic lupus erythmatosis | 5.7 (2.0–16.2) |
| Nulliparity | 5.4 (2.8–10.3) |
| Maternal age |
3–4 (4.7–9.6) |
| Chronic hypertension | 3.8 (3.4–4.3) |
| Diabetes Mellitus | 3.6 (2.5–5.0) |
| Sister with preeclampsia | 3.3 (1.5–7.5) |
| Strong family history of cardiovascular disease | 3.2 (1.4–7.7) |
| Twin pregnancy | 3 (2–4.2) |
| COVID-19 | 2.84 (1.67–4.82) |
| Obesity | 2.5 (1.7–3.7) |
| Body mass index |
2.3–2.7 (2.8–4.4) |
| Previous partner had PE | 1.8 (1.6–3.9) |
| Excessive weight gain | 1.7–2.8 (1.9–4.4) |
| Artificial insemination | 1.6–2.5 (1.7–2.9) |
| Use of contraceptive barrier method | 1.6–2.1 (4.3–7.3) |
| Pregnant woman born with low birth weight | 1.4 (1.6–3.0) |
| Bleeding in 1st trimester | 1.4 (1.4–2.6) |
Preeclampsia progresses in 2 stages: in the first stage, uterine spiral artery remodeling disordered, resulting in abnormal trophoblastic infiltration and superficial placenta implantation in the maternal myometrium. In the second stage, some maternal syndrome appears, which is associated with the systemic inflammatory response caused by the placenta, leading to the signs of preeclampsia [3, 6]. Delivery can resolve most signs and symptoms; however, preeclampsia can persist after delivery and recurrence in the postpartum period [3].
At present, the pathogenesis of preeclampsia is still unclear, large number of hypothesis have been proposed for the placental dysfunction, including oxidative stress, abnormal natural killer cells at the maternal-fetal interface, and genetic and environmental factors, though none have conclusive evidence in humans [6]. This article summarized the research status of pathogenesis in preeclampsia as follows (Fig. 1).
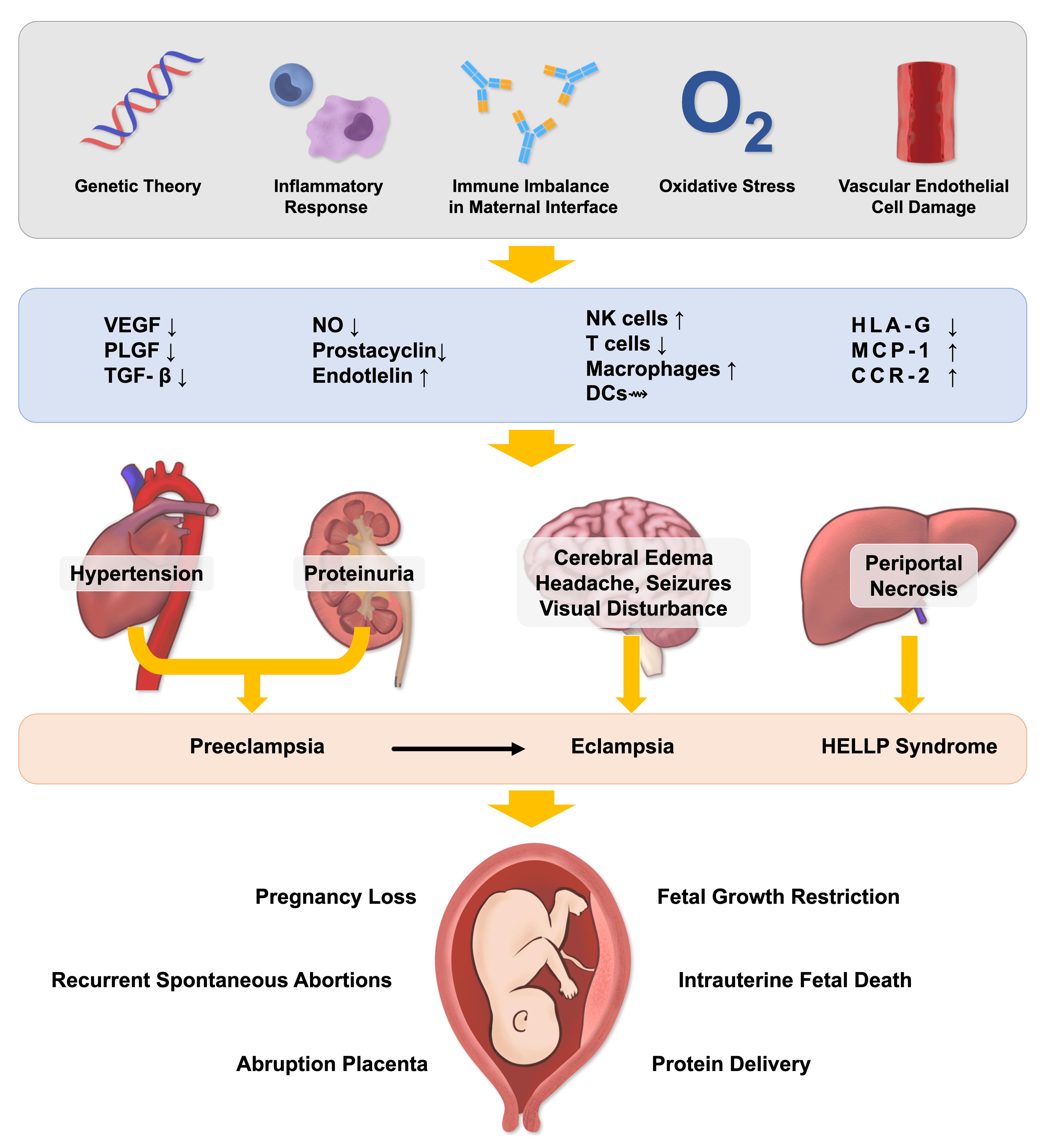 Fig. 1.
Fig. 1.Mechanism of preeclampsia.
Family history is an independent risk factor of preeclampsia [7]. As early as the 1870s, the literature reported a family gathering phenomenon of preeclampsia [8]. The preeclampsia incidence of pregnant women can be increased by 2 to 5 times, whose immediate family members suffered with preeclampsia previously [9, 10]. A family history of preeclampsia can significantly increase the risk of individual women suffering from preeclampsia by 24% to 163% [11]. A study from Norway showed that women born with preeclampsia has a 2.2-fold increased risk of preeclampsia during her pregnancy, indicating that preeclampsia was genetic predisposition [12].
For men whose mothers had preeclampsia, the men might also increase the incidence of preeclampsia in their spouses [13]. Dekker [14] claimed that, for the husbands, if their original spouses had a history of preeclampsia, when they had a next spouses without preeclampsia, the probability of preeclampsia in the new one would also increase significantly. Nearly half of preeclampsia family aggregation is caused by maternal genetic factors; and environmental, fetal or paternal factors account for the rest [15]. Nowadays, a variety of genetic models have been proposed to explain the inheritance of preeclampsia, including classic Mendelian Inheritance models [16], mitochondrial genes [17]. However, no hypothesis can fully explain preeclampsia. Therefore, preeclampsia is thought to be the result of complex interactions between maternal and fetal genotypes in addition to the environment [18].
Compared with healthy pregnant women, the placenta of preeclampsia patients often present vascular abnormalities and inflammation, leading the reduction of placental spiral artery remodeling and increasing vasoconstriction [19]. Macrophages, natural killer cells (NK), dendritic cells (DCs), and T cells infiltrate in the decidua around the trophoblast cells, controlling the remove of inherent cells in the spiral artery and the invasion of trophoblasts [20, 21, 22]. Any imbalance of these local inflammatory reactions may lead to placental malformations, impairing placental blood supply, resulting in preeclampsia, urine protein and edema, and even pregnancy loss [23].
Uterine Natural Killer (uNK) cells are the main lymphocytes infiltrating the decidua, accounting for about 70% of the immune cells at the maternal-fetal interface and regulating the depth of trophoblast cell infiltration and angiogenesis, playing a role in the abnormal placentation observed in preeclampsia [18, 24]. Unlike peripheral NKs, uNK is not cytotoxic [18]. The uNK cells in the preeclampsia were significantly higher than those in the normal pregnancy group between 12 and 20 weeks of pregnancy [25].
There are specific receptors, such as killer cell immunoglobulin-like receptors (KIRs) on the surface of uNK cells in the maternal-fetal interface, which are related to the onset of preeclampsia [26]. The HLA-G of trophoblasts can contact with KIRs to regulate the invasion ability of trophoblasts [24, 27]. However, when the uNK cells express the KIR inhibitory, the trophoblasts will tend to express HLA-C2, which will inhibit the function of NK cells in the maternal-fetal interface, and the resistance to spiral artery remodeling will increase significantly [28]. Uterine NK cells also mediate immune tolerance by recognizing major histocompatibility complex (MHC) ligands of fetal trophoblast cells [24].
T lymphocytes play an important role in mediating the occurrence of preeclampsia [29]. The imbalance of T cells, appearing in the blood circulation of women with low placental function, leads to a chronic inflammatory state, which is believed to be one of the causes of the disease [30].
During a normal pregnancy, maternal Th1 and Th2 establish a new balance, when
the immune state drifts to Th2, the Th1 cytokines are down-regulated, promoting
the normal placenta development and avoiding immune rejection of embryos in utero
[31, 32]. Th1 cells can secrete interleukin-2 (IL-2), tumor necrosis factor
(TNF)-
Both Th1 and Th2 cells were differentiated from Th0 cells [32]. This differentiation of Th0 cells is regulated by regulatory T cells (Treg cells) [31, 34]. Treg cells play an anti-inflammatory effect and the main effector cytokine is IL-10, helping form immune tolerance and hinder the differentiation [31]. During this differentiation process, specific transcription factors GATA-3 promotes Th0 cells to differentiate into Th2 cells, and T-bet induces Th0 cells to differentiate into Th1 cells [35].
In preeclampsia patients, the levels of Treg and indoleamine 2,3-dioxygenase (IDO) are significantly lower than that in normal pregnant women [36]. IDO is an amino acid converting enzyme, which can effectively regulate Treg activity. When IDO decreases, Tregs activity will also decrease, promoting the transformation of Th0 cells to Th17 cells [37, 38]. IL-17 is a pro-inflammatory factor (most of the core cytokines secreted by Th17), which can promote inflammation. Immune imbalance between Th17 cells and Treg cells is one of the important causes of preeclampsia [39].
In the process of human embryo implantation, extra-villous trophoblasts (EVT) derived from blastocysts invades the decidua, interacting with natural killer cells, enhancing the expression of angiogenic factors and microangiogenesis [40], increasing blood flow in the uterus ang and promoting the development of the fetus and placenta [41].
In early pregnancy, uterine macrophages participate in the initiation apoptosis of vascular smooth muscle cells and endothelial cells during decidual vascular remodeling [42]. The macrophage polarization imbalance damages trophoblast and placental function, and leads to preeclampsia, FGR, miscarriage and even preterm delivery [43]. Colony stimulating factor (CSF) is an effective inducer of macrophage proliferation, differentiation and activation [44]. Serum CSF in pregnant women are elevated and quickly return to baseline levels after the delivery [44]. The dynamic phenotypic changes of macrophages indicate that: (1) The macrophage expression is elevated in decidua during preeclampsia [45]. (2) preeclampsia significantly enhances the expression of CSF in early decidual cells [46].
In late pregnancy, the elevated macrophage and loss of trophoblast were observed under some abnormal pregnancy conditions, including early pregnancy loss, preeclampsia, FGR and gestational trophoblastic disease, indicating macrophages homeostasis is essential for maintaining decidual homeostasis in human pregnancy [47, 48, 49].
Th1 and Treg cells in the placenta and decidua are differentiated from naive CD4+ T cells induced by antigen presenting cells (APCs), the most important of which are dendritic cells (DCs) [50]. Low-level stimulatory molecules expressed by immature DCs (such as MHC-II and CD80/CD86, etc.) can induce naive CD4+ T cells to differentiate into Treg cells and form immune tolerance [51]. While mature DCs express high levels of stimulatory molecules would induce naive CD4+ T cells to differentiate into activated Th1 cells and mediate inflammation [52]. During a normal pregnancy, the number of DCs at the maternal-fetal interface increases and maintain immature [52, 53]. Between the patients with preeclampsia and the normal pregnant patients, the number of DCs in the decidua is no significant difference; however, the ability of DCs inducing Treg cell is significantly impaired in patients with preeclampsia, and the mechanism is still not sure [51].
Pregnancy can be considered as an immune-homeostasis between the pregnant woman’s tolerance and the protection of the fetus [54]. Normal pregnancy is a state that immune system is activated and pregnancies affected by preeclampsia are regarded to involve more complex immunological processes. The boundary between fetal components and maternal tissues is the maternal-fetal interface. The balance of maternal-fetal immune regulation is the vital to successful pregnancy [55]. In normal pregnancy, there is an increase in systemic immune cell activation, especially monocyte (the macrophage precursor) [43]. When infiltrated into tissues, circulating monocytes will differentiate to steady state macrophages [23]. Both of monocytes and macrophages are related to trophoblast growth, spiral artery remodeling and angiogenesis [56]. Among the pregnant women with preeclampsia, the monocyte chemoattractant protein 1 (MCP-1), mRNA of MCP-1 and C-C chemokine receptors 2 (CCR2, the receptor of MCP-1) in serum and placental tissue are significantly increased [57]. It is believed MCP-1 may induce excessive local placental inflammation by binding to its receptor CCR2, disrupting the immune balance and causing preeclampsia [57].
For pregnant women with preeclampsia, after losing the immunity homeostasis, the immunity to tolerate fetal antigens would decrease, causing abnormal placental immune function, inducing inflammatory reactions and inhibiting blood vessel production [58]. In addition, DCs in the decidua also induce mother’s immune tolerance to the fetus by promoting Th2 cells in uterus and placenta [36]. In addition, the appropriate concentration of human leukocyte antigen G (HLA-G) can protect the embryo from the attack by cytotoxic T lymphocytes (CTL) [59]. Johnsen showed that when the expression of HLA-G in trophoblast cells is reduced, it is vulnerable to maternal immune cells, which can lead to preeclampsia, miscarriage or FGR [60].
Oxidative stress (OS) is defined as an imbalance between oxidants and antioxidants, leading to a disruption of redox signaling and control and/or molecular damage [61, 62]. The placenta is hypoxic in the first trimester of pregnancy, and the blood flows through the villus space in the later period and releases a large amount of reactive oxygen species (ROS), with an increase of antioxidants in the placenta to protect pregnant women and fetus from oxidative stress outbreaks [63].
Preeclampsia is associated with insufficient trophoblast infiltration and impaired vascular remodeling in spiral arteries [64]. Impaired vascular remodeling in preeclampsia reduces placental perfusion and oxygenation, and increases oxidative stress with ROS and toxic lipid peroxides [65]. Free radicals activate monocytes and neutrophils to produce pro-inflammatory cytokines, and further produce ROS through the action of various enzymes and the large amount of ROS produced by oxidative stress can damage the trophoblast cell mitochondrial membrane, permeable transport pore, mitochondrial DNA, and mitochondrial enzymes, which in turn triggers preeclampsia, premature rupture of membrane, etc. [63]. The expression of ROS damaging the vascular endothelium is significantly increased in the blood of women with preeclampsia; while the concentration of antioxidants weaken the activity of ROS in preeclampsia is significantly reduced [66, 67].
In preeclampsia women, the maternal immune response, against the fetus, interferes with trophoblast invasion and angiogenesis, resulting in oxidative stress and a maternal systemic inflammatory response [68]. Immunity imbalance of Th1/Th2 cells, Th17/Treg cells and abnormal regulation of decidual NK cells will lead to the failure of trophoblast invasion and the reduction of placental perfusion, which would generate of reactive oxygen species, and the aggravate of oxidative stress Autoreactive antibodies to the angiotensin receptor AT1 are elevated in preeclamptic mothers and inhibit trophoblast invasion, thereby promoting oxidative stress [69]. Complement system also plays a crucial role in maintaining a healthy pregnancy, and abnormal activation of the complement system would cause maternal systemic inflammatory response, leading to the occurrence of preeclampsia.
Malonyldialdehyde (MDA) is a lipid peroxide, which is an important monitoring indicator of oxidative stress [70]. When the placental ischemia and hypoxia occur, the oxidative stress is intensified and releases a large amount of oxygen free radicals, which penetrate the cell membrane and attack the polyunsaturated fatty acids in the cell, converting them into lipid peroxides, and MDA as the product of lipid peroxidation reaction will increase significantly [71, 72].
8-isoprostane is another final product of lipid peroxidation, catalyzed by arachidonic acid through oxygen free radicals [73]. Its level is consistent with the change trend of lipid peroxidation, which could be an indicator of lipid peroxidation reaction [74]. The more severe preeclampsia, the significantly higher 8-isoprostane in serum; which indicated the oxidative stress in preeclampsia is correlated with the severity of the disease [75].
Patients with preeclampsia got mitochondrial dysfunction, increased mitochondrial lipid peroxidation, and susceptibility to oxides [76]. In mitochondria of women with preeclampsia, those genes responsible for energy production and oxidative electron exchange, such as cytochrome C oxidase, are abnormally expressed [77]. Compared with normal pregnancy, endothelial cells treated with plasma from women with preeclampsia had significantly higher intramitochondrial ROS, inhibiting cell death ROS-induced [78].
The pathological mechanisms of preeclampsia include insufficient placental bed vascular remodeling, placental ischemia and hypoxia, secondary to systemic endothelial injury [16]. It is currently believed that the increase in the adhesion of leukocytes to endothelial cells and the endothelial cell permeability mediated by oxygen free radicals are critical processes in the development of preeclampsia [24, 79].
Vascular endothelial growth factor (VEGF) can enhance vascular permeability, increase the ability of neovascular endothelial division and proliferation, regulate the permeability of placental vascular endothelial cells and promote blood vessels formation through the mediation of nitric oxide (NO) [74]. When the vascular endothelium is damaged, VEGF levels and the function of placental vascular endothelial cells will decrease, involving the occurrence of preeclampsia [80]. The levels of VEGF protein and mRNA in placental tissues of pregnant women with preeclampsia were lower than those of normal pregnant women [81]. As pregnancy progressed, the density of micro-vessel in normal placental were increased, while decreased in placental with preeclampsia, indicating VEGF inhibited the occurrence of preeclampsia [82]. The VEGF level in placenta preeclampsia was related to the uterine spiral artery resistance index and systolic period of pregnant women with preeclampsia, indicating the lower the serum VEGF level in pregnant women with preeclampsia, the greater blood flow resistance, and the less in placental perfusion [83].
Also, vascular endothelial growth factor or placental growth factor (PlGF) and anti-angiogenic factors like soluble fms-like tyrosine kinase 1 (sFlt-1) are known to be related to the disease pathogenesis [84]. Recombinant sFlt-1 could block development of endothelial tubes and inhibit vasodilatory effects of VEGF and PlGF in vasculature [85]. In preeclampsia, circulating maternal serum levels of sFlt-1are increased, and PlGF levels are decreased [86].
In addition, endothelial cell damage can promote the release of tissue factor (TF), plasminogen activator inhibitor (PAI), thrombomodulin, platelets, etc., and activate the coagulation system [87]. The expression of TF in pregnant patients with hypertension was significantly higher than that in normal pregnant women. It was believed that the change of TF may cause abnormal coagulation function in pregnant women, promoting the occurrence of preeclampsia consequently [88]. Also, PAI elevated in pregnant women with preeclampsia [89, 90].
Thrombomodulin is a transmembrane glycoprotein existing in vascular endothelial cells and trophoblast cells. Thrombomodulin is the receptor for thrombin, which can bind to thrombin to form a thrombomodulin-thrombin complex [91]. This substance has an activating effect on protein C form and can activate it into protein C, which can protect endothelial cells, reduce inflammation, anti-thrombosis, improve microcirculation in turn [92, 93].
When the expression of thrombomodulin in the placenta tissue is reduced, the inflammatory response is prone to over-activation, damaging vascular endothelial cells [93, 94]. A lower expression of thrombomodulin in the placental tissue of pregnant women may be related to the evolution of early-onset severe preeclampsia [94]. Platelet may promote of blood coagulation and mediate inflammation in the occurrence and development of preeclampsia [95].
Nitric Oxide (NO) is one of the key endothelial release factors in the body and the primary neurotransmitter; playing a role in regulating vascular blood flow, arterial blood pressure, maternal organ perfusion and placental blood flow, maintaining the metabolism, and keeping dynamic balance of blood pressure in cardiovascular [96].
In vascular endothelium, nitric oxide synthase (NOS) is the rate-limiting enzyme
in NO synthesis, and the expression of NOS depends on the regulation of nuclear
factor kappa-B (NF-
In vascular smooth muscle cells, NO can effect on cyclic guanosine phosphate, causing calcium outflow, promoting the conversion of free calcium into bound calcium and vasodilation [100]. Blocking NO synthesis with NOS inhibitors in pregnant rats would cause preeclampsia-like symptoms, including hypertension, proteinuria, FGR, even stillbirths and malformations [100, 101]. In pregnant mice with NOS knockout, the uterine blood flow were reduced and spiral arteries were shortened, leading placenta oxygen supply insufficient [102, 103]. The reduction of NO may cause an imbalance of vascular factors, followed by arteriole spasm and increased peripheral resistance, especially in kidney, uterus and placenta, which promotes the development of hypertension and preeclampsia [104]. NO is the important link in vascular endothelial cell damage and oxidative stress, which can ensure the blood supply of the placenta and the nutrition and oxygen supply of the fetus [105].
IVF is associated with the onset and progression of PE. Defective placentation and placental insufficiency may predispose IVF patients to preeclampsia and may manifest as first-trimester bleeding [106]. In IVF protocols including oocyte donation, the absence of corpus luteum and subsequent deficiency of relaxin can disturb maternal circulation and precipitate the development of preeclampsia [107]. Oocyte donation is an independent risk factor for preeclampsia due to immunological maladaptation or intolerance [108].
Among women with pregestational diabetes, preeclampsia complicates 10% to 20% of the pregnancies [109]. GDM and preeclampsia share many risk factors, including advanced maternal age, nulliparity, multifetal pregnancies, non-white race/ethnicity and pre-pregnancy obesity [110].
Coronavirus disease 2019 (COVID-19) has been shown to cause systemic complications such as high blood pressure, kidney disease, thrombocytopenia, and liver injury [111]. COVID-19 during pregnancy is strongly associated with preeclampsia. COVID-19 severity does not seem to be a factor in this association [112]. Women with preeclampsia should be considered a particularly vulnerable group with regard to the risks posed by COVID-19 [111].
Preeclampsia is a serious complication during pregnancy, which greatly threatens the pregnant women and fetuses. The pathogenesis of preeclampsia is still unclear, the theory of Genetic, Inflammatory Response, Immune Imbalance in Maternal-Fetal Interface, Oxidative Stress, Vascular Endothelial Cell Damage are supposed involved in the progress of preeclampsia. Although there are various theories mentioned above, none of them can fully explain all the biological behaviors of preeclampsia. More research is needed on the mechanism of preeclampsia.
RL—Project development, Literature Collection, Manuscript writing. BZ—Literature Collection, Critical revision of the manuscript, Supervision. XZ—Project development, Literature Collection, Critical revision of the manuscript, Supervision. All authors read and approved the final manuscript.
Not applicable.
We would like to express our gratitude to all those who helped us during the writing of this manuscript. Thanks to all the peer reviewers for their opinions and suggestions.
This research received no external funding.
The authors declare no conflict of interest.
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.