
- Academic Editor
Ischemia-reperfusion injury (IRI) is a complex phenomenon. Although researchers have long been aware of IRI, its complex signaling events and potential therapeutic targets are still an active research area. The role of reactive oxygen species in IRI has garnered great interest among scientists. Recent studies have found that reactive oxygen species produced by IRI can activate redox-sensitive transient receptor potential channels (redox TRPs). The discovery of redox TRPs provides a new perspective for understanding the mechanism of IRI.
When a tissue or organ is in a state of hypoperfusion, early restoration of blood supply is a priority. However, the reperfusion process itself activates cellular events such as inflammatory responses, autophagy, and cell death programs, which worsen tissue damage [1]. The phenomenon in which restoration of perfusion and oxygen supply to certain ischemic tissues and organs aggravates the injury instead is called ischemia-reperfusion injury (IRI). Using isolated cells to simulate reperfusion injury, it was found that more than 90% of cell death occurs during reperfusion [2]. Hearse et al. [3] also observed this phenomenon in reperfused cardiac tissue. The concept of IRI has received increasing attention and has thus opened up a new field of scientific research. IRI has been described as one of the primary factors contributing to the observed disability and mortality rates in various clinical entities, including myocardial infarction (MI) [4], cardiac arrest [5], ischemic stroke [6], and acute kidney injury (AKI) [7]. Although IRI is not new, the complex signaling events induced and multitargeted therapies are still hot topics. Therapeutic options for IRI have extended from traditional drug therapy to targeted delivery and controlled release using nanoparticle drug delivery systems [8, 9, 10].
Transient receptor potential (TRP) channels, which were originally discovered in Drosophila melanogaster, are a superfamily of cation channels with six putative transmembrane segments [11]. They have received much attention in recent years for their different roles in various physiological processes. TRP channels are highly conserved among species. Mammalian TRP channels are divided into six subfamilies based on amino acid sequence homology: TRP canonical, TRP vanilloid (TRPV), TRP melastatin (TRPM), TRP ankyrin (TRPA), TRP polycystic, and TRP mucolipin [12]. TRP channels have a tetrameric structure assembled from four protein subunits. Each subunit of TRPs consists of six transmembrane regions and a pore-forming region (between the fifth and sixth transmembrane regions), with the C- and N-termini facing the cytoplasm of the cell. The different ways in which the subunits are combined determine the TRP channel type. Bivalent cation channels TRPs have multimodal sensing capabilities that translate a wide range of stimuli (e.g., thermal and cold stimuli, osmotic pressure, mechanical stress, and cellular redox states) into specific cellular responses [13]. Among them, a newly identified group of TRP channels regulated by the cellular redox state is called redox-sensitive TRP channels (redox TRPs). Redox TRPs (including TRPM2, TRPV1, and TRPA1) are sensitive to reactive oxygen species (ROS) and are activated by oxidative products either directly or indirectly [14]. TRPA1 is a non-selective cation channel present in various tissues and organs and is mainly expressed in sensory neurons. Activation of TRPA1 is closely related to cold sensation generation and transmission as well as the regulation of pain and inflammation [15]. TRPV1 is widely distributed in the peripheral nervous system; when activated, it can produce pain, burning and itching sensations, and cause the release of pro-inflammatory mediators such as substance P and calcitonin gene-related peptide (CGRP). Thus, both TRPA1 and TRPV1 are widely involved in neuropathic pain, inflammatory pain, and pruritus; however, the concentration of endogenous agonists required to activate TRPV1 channels is much higher than that of TRPA1 [16, 17]. TRPM2 also functions as a temperature sensor in neurons [18]. More importantly, TRPM2 acts as a bioreceptor for oxidative stress (OS) in tissues such as the brain, heart, and blood vessels, regardless of physiological or pathological conditions [19].
One of the key mechanisms of IRI is OS. When blood flow is restored to ischemic tissues, ROS such as superoxide anions and hydrogen peroxide are produced. These ROS can directly damage cellular macromolecules, including lipids, proteins, and nucleic acids, leading to cellular dysfunction and death. Therefore, targeting OS is an important therapeutic strategy to mitigate IRI [20]. In this context, redox TRPs have emerged as potential factors in the pathogenesis of IRI disease. This close association has also inspired research trends [21].
In this article, we focus on the mechanism of action and pharmacological value of TRPM2, TRPV1, and TRPA1 in IRI.
The concept of OS, first introduced in 1985, refers to the production of excessive amounts of highly reactive molecules, such as ROS and reactive nitrogen radicals (RNS), in the body during the removal of aging cells or when exposed to a noxious stimulus, which results in oxidative processes that exceed the body’s ability to clean up, triggering tissue damage [22, 23]. ROS include not only oxygen radicals (e.g., superoxide anion, hydroxyl radical) but also some oxygen-containing non-radical derivatives, such as monoclinic oxygen and hydroperoxides. By detecting an increase of ROS in ischemic tissue cells [24] and accelerating the clearance of ROS, the increase of blood–brain barrier permeability after IRI [25] and MI area [26] can be alleviated, all of which confirm the key role of ROS in IRI.
In IRI, there are six main mechanisms involved in the increase of ROS. (1) Impaired mitochondrial function: mitochondria are a major source of ROS, especially during IRI. In ischemia, electrons in the electron transport chain spill over to oxygen, generating superoxide and ROS [27]. After reperfusion, ROS increases dramatically, exacerbating mitochondrial dysfunction and creating a vicious cycle. (2) Xanthine oxidase (XO): xanthine dehydrogenase is the predominant form of xanthine oxidoreductase in normal healthy tissues, but is affected by calcium overload and reduced ATP synthesis, and is converted to XO by calcium-dependent proteases [28]. In addition, the catabolism of ATP leads to the accumulation of hypoxanthine, and the accumulated hypoxanthine is sequentially converted to xanthine and uric acid by the action of XO, accompanied by the production of oxygen free radicals [29]. (3) (Reduced) nicotinamide adenine dinucleotide phosphate (NADPH) oxidase system: phagocytic activity significantly increases the oxygen consumption of phagocytes (especially neutrophils), catalyzed by the intracellular NADPH/nicotinamide adenine dinucleotide phosphate (NADP) enzyme system, and the oxygen taken up accepts electrons to form ROS [30, 31]. (4) Catecholamine autooxidation: IRI leads to excessive production of catecholamines and their oxides, accompanied by the generation of large amounts of oxygen radicals [32]. (5) Nitric oxide synthase (NOS): uncoupled NOS is an important source of ROS. IRI is associated with reduced bioavailability of NOS cofactors and NOS uncoupling [33]. (6) Decreased antioxidant system: overproduction of ROS leads to OS, but the body has antioxidant systems to scavenge ROS including superoxide dismutase, catalase, and glutathione peroxidase. These systems are essential to maintain normal cellular function [34].
IRI is characterized by intracellular calcium overload and ROS production.
Approximately 44% of free intracellular calcium is stored in the mitochondria
and endoplasmic reticulum and is dependent on calcium pumps
(Ca
OS is a physiological process triggered by the excessive accumulation of free radicals and oxidants in the body and often involves oxidative modification of proteins, lipids, and DNA. Although cysteine (Cys) is one of the least abundant amino acids, it is the catalytically active center of many enzymes and plays a crucial role in maintaining protein function. During OS, Cys is often the target of ROS [39]. The sulfhydryl group (-SH) of the cysteine side chain can be oxidized by ROS to sulfinic acid (-SOH), and then further oxidized to sulfinic acid (-SO2H) and sulfonic acid (-SO3H) [40]. In conclusion, many TRP channels sense changes in cellular redox status by utilizing Cys residues.
ROS and lipid peroxidation products such as 4-hydroxynonenal and
15-deoxy-
TRPM channels consist of an N-terminal, C-terminal, and channel structural
domain. Among them, the homology between the NUDT9-H domain at the C-terminus of
TRPM2 and NUDT9 ADP-ribose hydrolase makes TRPM2 a novel protein with the dual
functions of an ion channel and hydrolysis of ADP-ribose [47]. TRPM2 is a
non-selective calcium-permeable cation channel and functions as a bridge between
ROS and calcium [48]. When the cellular antioxidant system fails to effectively
scavenge ROS and other free radicals, a potentially deleterious cascade of events
occurs within the cell, namely, excess ROS leads to DNA strand breaks, which in
turn trigger the activation of two key enzymes: poly(ADP-ribose) polymerases
(PARPs) and poly(ADP-ribose) glycohydrolases (PARGs). PARPs and PARGs mediate the
sequential conversion of NAD
In IRI, activation of ROS-sensitive TRP channels (especially TRPM2 channels) is mainly driven by pathological increases in intracellular calcium ions and overproduction of ROS, which in turn trigger a wide variety of cellular signaling pathways leading to tissue damage and dysfunction Therefore, the study of redox TRPs and their modulators is of great significance (refer to Fig. 1).
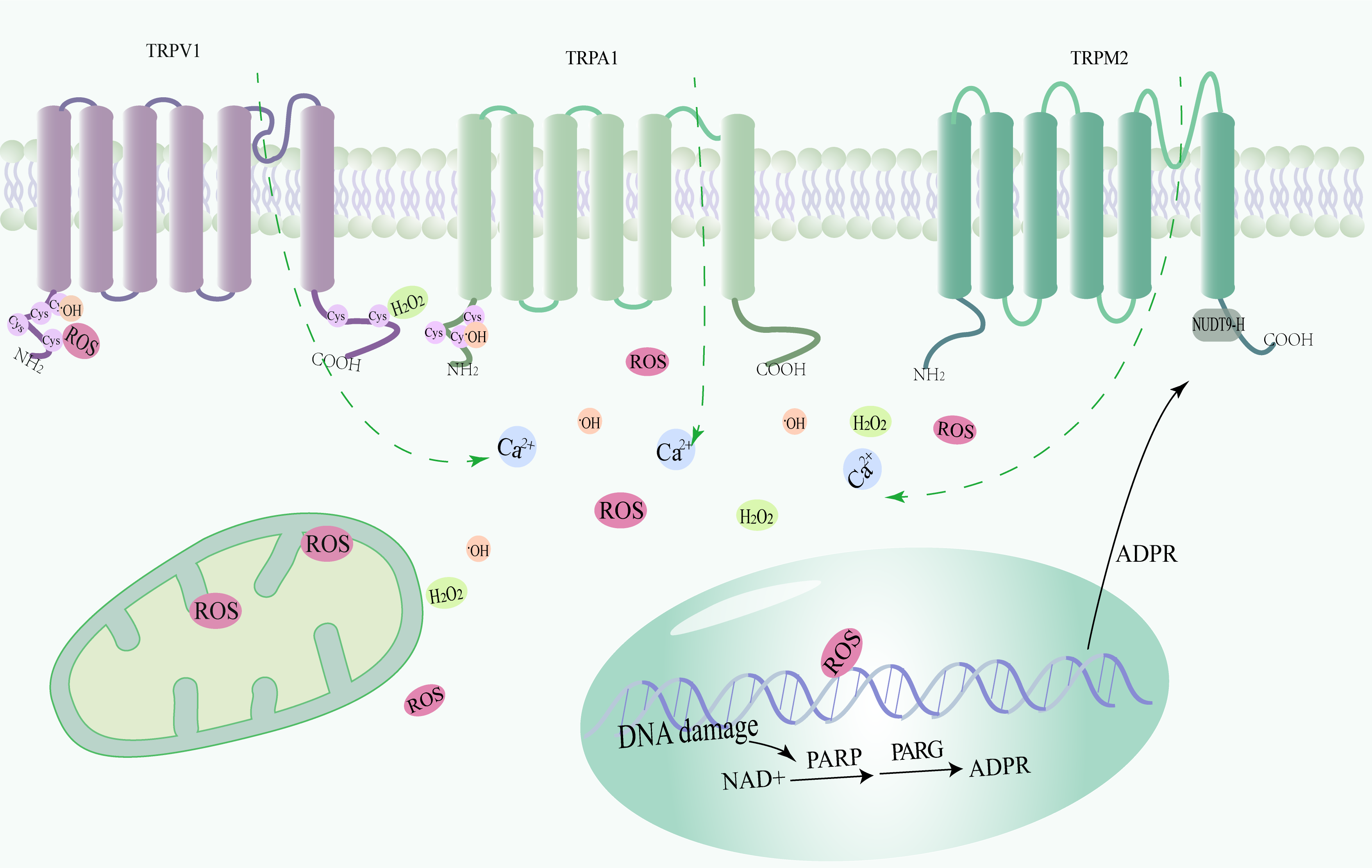 Fig. 1.
Fig. 1.Structure and activation mechanism of redox-sensitive transient receptor potential (TRP) channels (TRPV1, TRPA1, and TRPM2). Transient receptor potential (TRP) channels sensitive to redox reactions trigger calcium inward flow after sensing excess reactive oxygen species. Sequential activation of poly (ADP-ribose) polymerases (PARPs) and poly (ADP-ribose) glycohydrolases (PARGs) mediates the conversion of nicotinamide adenine dinucleotide (NAD) to ADP-ribose. Subsequently, calcium inward flow through TRPM2 channels was triggered. ROS, Reactive oxygen species.
After ischemia, when blood reenters the tissues, it triggers the release of large quantities of oxygen free radicals. In turn, these free radicals cause oxidative damage to the cell membranes and the production of toxic metabolites, ultimately leading to cellular DNA, protein, and lipid damage. The effects of IRI extend far beyond direct tissue damage as major organs are also affected including the heart, brain, liver, and kidneys [50]. We review the role and mechanism of redox TRP channels in several types of IRI-induced organ damage (Refer to Table 1, Ref. [51, 52, 53, 54, 55, 56, 57, 58, 59, 60, 61, 62, 63, 64, 65, 66]).
| Redox TRP | Cell lines/animals/human used in redox TRP studies | Effect | References |
| TRPA1 | Cardiac fibroblasts | Involved in cellular phenotypic transition. | [51] |
| Heterozygous Trpa1 |
Trpa1 knockdown exacerbates RIRI with classical activation of macrophages. | [60] | |
| Human kidney-2 cells, Male Trpa1 |
Promotes transcription of inflammatory factors. | [61] | |
| TRPV1 | Male TRPV1 |
TRPV1 activation alleviates cardiac IRI. | [53] |
| Male Sprague-Dawley rats, DRG neurons | Inhibition of TRPV1 upregulation in dorsal root ganglia after MI contributes to exogenous cardioprotection. | [52] | |
| B6.129X1-TRPV1 KO mice, astrocytes | TRPV1 mediates astrocyte activation and IL-1 |
[56] | |
| Male TRPV1 |
TRPV1 protects RIRI in diet-induced obese mice by enhancing CGRP release and increasing renal blood flow. | [62] | |
| Male TRPV1 |
KO of TRPV1 exacerbates IR-induced renal inflammation and injury in obese mice. | [63] | |
| TRPM2 | Human cardiomyocytes, human aortic endothelial cells, Wistar rats | Uric acid promotes MI injury via activating NLRP3 inflammasome and ROS/TRPM2 channel/calcium pathway. | [54] |
| PC12 cells | Knockdown of TRPM2 reduces apoptosis, OS, and NLRP3 inflammatory activation due to IRI. | [55] | |
| Human embryonic kidney 293 cells, male Trpm2 KO mice | Inhibition of TRPM2 exerts neuroprotective effects. | [57] | |
| Trpm2 C57BL/6 mice, human embryonic kidney293 cells | Aggravating calcium-dependent brain injury. | [58] | |
| TRPM2 |
Deficiency of the ROS-activated TRPM2 channel protects neurons from cerebral IRI by upregulating autophagy. | [59] | |
| Trpm2 KO C57BL/6 mice, human liver cell line L-02 | Downregulation of TRPM2 attenuates hepatic IRI through activation of autophagy and inhibition of the NLRP3 inflammasome pathway. | [64] | |
| Male C57BL/6 mice | Silencing of TRPM2 gene expression in mouse liver tissue attenuates HIRI. | [65] | |
| Trpm2 |
Inhibition of TRPM2 attenuates ferroptosis in HIRI. | [66] |
IRI, ischemia-reperfusion injury; RIRI, Renal IRI; KO, knockout; CGRP, calcitonin gene-related peptide; ROS, reactive oxygen species; MI, myocardial infarction; HIRI, Hepatic IRI.
Myocardial IRI (MIRI) is one of the leading causes of morbidity and mortality worldwide. This injury usually occurs postoperatively and can severely impair cardiac function. One of the serious complications is MI, which is one of the life-threatening pathologies in cardiac patients. One of the core treatments for MI is the relief of ischemia through prompt myocardial reperfusion; however, this process is accompanied by several challenges. In particular, reperfusion of ischemic tissue triggers an overproduction of ROS, which further aggravates myocardial injury. TRPA1 is expressed on both neurons and cardiomyocytes, but has no direct effect on cardiomyocytes [67]. It was found that sensory neurons co-cultured with cardiomyocytes in vitro may release protective substances to improve the survival rate of cardiomyocytes in the context of IRI [68]. Comparing the two genotypes of mice with similar basal cardiovascular function, the area of MI was significantly reduced in IRI-induced TRPA1-null mice [69]. Recent studies have shown that MI upregulates TRPA1 expression, increases intracellular calcium concentration and leads to calcineurin activation, triggering the translocation of the transcription factor nuclear factor of activated T cells (NFAT) to the nucleus to participate in the phenotypic transition of cardiac fibroblasts [51]. TRPV1 expression has also been found to be upregulated in myocardial ischemia [52]. Activation of TRPV1 by the protease-activated receptor 2 via the enzyme 12-lipoxygenase triggers a cascade of events, including the release of CGRP and substance P, two neuropeptides that play important roles in pain transmission and inflammation in MI [53]. Significant upregulation of TRPV1 protein expression and protein kinase A-mediated channel activity in dorsal root ganglion has been observed after IRI. Intrathecal morphine preconditioning significantly inhibits TRPV1 protein expression and phosphorylation, and reduces the MI area [52]. Previous studies have shown that ROS levels in TRPM2 knockout (KO) myocytes are significantly higher after hypoxic reoxygenation than in wild-type cardiomyocytes [70]. Calcium prevents IRI by reducing mitochondrial oxidants through the intracellular circulation of TRPM2 in the heart tissue, and systolic dysfunction in the TRPM2 KO heart is aggravated [71]. Uric acid levels are significantly elevated in patients with acute MI, indicating that hyperuricemia is not only associated with chronic kidney disease (CKD) but is also closely related to acute MI. One study showed that high levels of uric acid in cardiomyocytes modulate the ROS/TRPM2/calcium pathway via the nucleotide-binding domain (NBD)– and leucinerich repeat (LRR)–containing protein (NLR) family pyrin domain containing 3 (NLRP3) inflammatory vesicles [54]. ROS is a potent endogenous activator of TRPML1 channels, and thus, TRPML1 can also be viewed as a redox TRP. TRPML1 inhibits autophagic flux in MIRI by releasing lysosomal zinc ions and disrupting the fusion of autophagic vesicles and lysosomes [72] (Fig. 2).
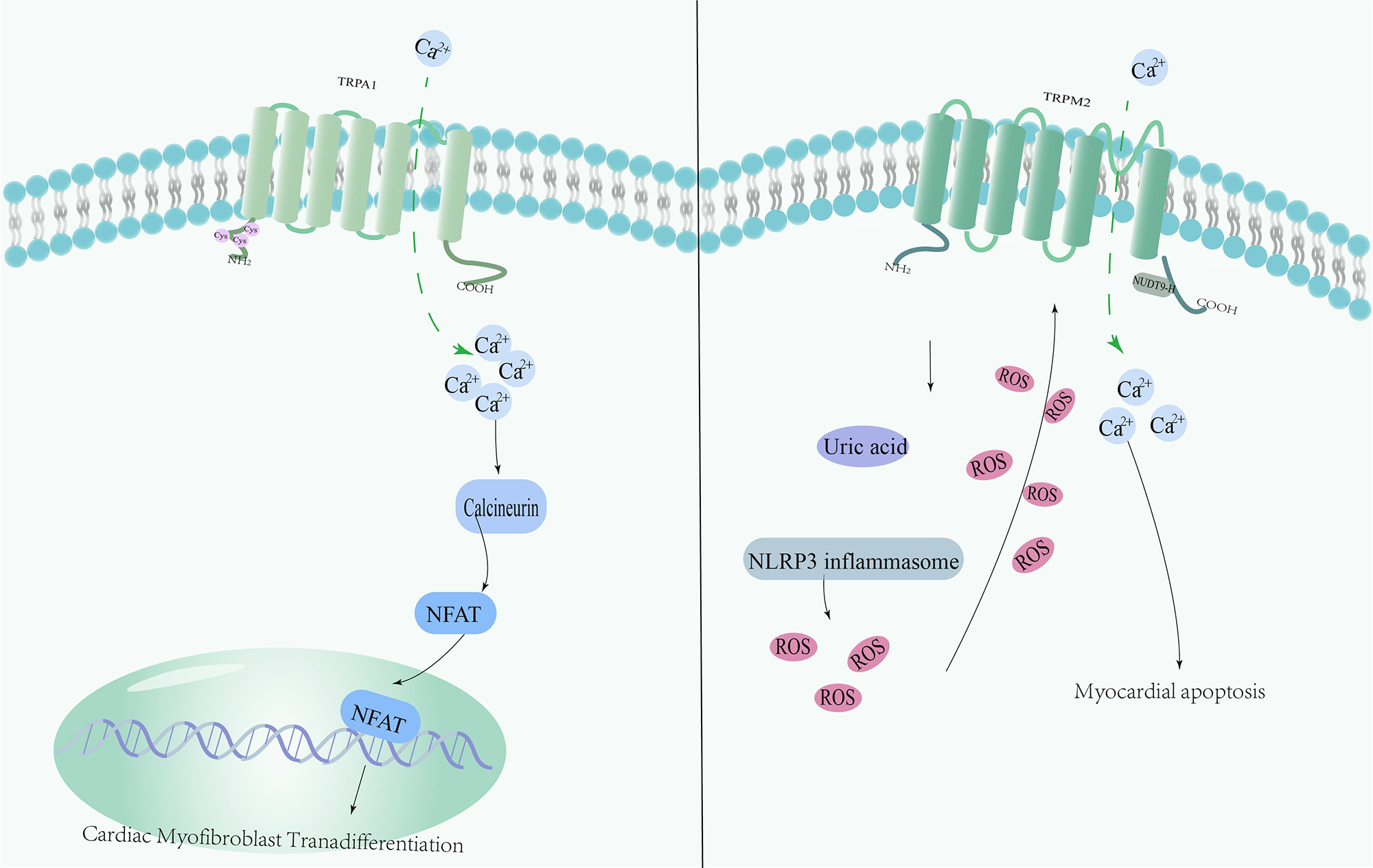 Fig. 2.
Fig. 2.Mechanism of redox-sensitive TRP channels involved in Myocardial IRI (MIRI). MI increases TRPA1 expression, leading to elevated intracellular calcium levels and activation of calmodulin. This in turn leads to translocation of nuclear factor of activated T cells(NFAT) to the nucleus, contributing to the phenotypic transformation of cardiac fibroblasts. Uric acid is elevated in patients with MI, and high levels of uric acid modulate the ROS/TRPM2/calcium pathway via NLRP3 inflammatory vesicles in cardiomyocytes.
Cerebral IRI (CIRI) occurs as a results of ischemic stroke. Stroke, especially
ischemic stroke, is the second leading cause of death and disability worldwide
[73]. Treatment of stroke or other cerebral ischemic diseases requires measures
to reduce reperfusion injury in addition to restoring blood flow as rapidly as
possible. Reperfusion may lead to the excessive production of oxygen free
radicals, a surge in the inflammatory response, disruption of the blood–brain
barrier, and abnormalities in neurotransmitter release. Accumulation of ROS
triggers apoptosis and other forms of cell death in neuronal cells, making OS
damage a major concern during the treatment of ischemic stroke [74]. TRPV1
regulates OS and apoptosis in microglia, which directly affect neuroprotection
and recovery after stroke [75]. PC12 cells are a commonly used cellular model in
neuronal cell research. Oxygen-glucose deprivation (OGD) is an experimental
method that mimics ischemic conditions and can contribute to ROS production.
Inhibition of TRPM2 channels may reduce OGD-induced neuronal apoptosis, OS, and
mitochondrial damage by decreasing the activation of NLRP3 inflammatory vesicles
[55]. Similar to TRPM2 channels, activation of TRPV1 channels is also closely
related to ROS production. Investigators found that in astrocytes cultured under
OGD conditions, TRPV1 signals through Janus kinase 2 (JAK2)/signal transducer and
activator of transcription 3 (STAT3) to regulate the expression of target genes
(glial fibrillary acidic protein and interleukin 1 beta [IL-1
In recent years, researchers have extensively studied the role of TRPM2 channels in CIRI. TRPM2 is a calcium channel that is widely expressed in the brain, including neurons, microglia, and macrophages [78, 79]. ROS not only triggers OS but also activates TRPM2 channels. Overactivation of TRPM2 channels further increases intracellular calcium concentration, creating a vicious cycle that leads to more cellular damage [80]. Bilirubin, a compound produced during the metabolism of hemoglobin, is known for its antioxidant properties, neutralizing ROS and protecting cells from OS. ROS, represented by hydrogen peroxide, is produced in large quantities in brain IRI, which sequentially undergoes the Fenton reaction and the action of PARP1 and PARG to generate ADPR, which is used to stimulate TRPM2 and mediate neuronal cell death [57]. Recent studies have revealed novel mechanisms by which bilirubin regulates TRPM2 channel activity, which provide interesting insights into its dual role within the cell. Researchers first used Spearman correlation analysis to show that elevated bilirubin may be a consequence of stroke, and that elevated bilirubin levels are positively correlated with infarct volume. Subsequently, it was revealed that bilirubin exacerbates ischemic brain damage by activating TRPM2 through binding to the cavity in the transmembrane region near the calcium-binding site of the channel [58]. ROS have been widely recognized as inducers of autophagy. However, in the pathological state of OS, ROS-TRPM2-calcium influx activates downstream calcium-calmodulin dependent protein kinase 2 and subsequently phosphorylates Beclin1 to inhibit autophagy [81]. There was no significant difference in Beclin1 (S295) phosphorylation level between wild-type mice and TRPM2 KO mice when brain tissue was subjected to IRI, whereas there was significant difference in the phosphorylation levels of AMPK and its downstream mammalian target of rapamycin (mTOR), which are also involved in autophagy regulation. TRPM2 activated by ROS downregulates autophagy through the AMPK/mTOR pathway, thus weakening the cellular self-protection mechanism and more easily leading to neuronal death [59]. The antioxidant N-acetyl cysteine inhibits the activation of TRPM2 with neuroprotective effects [82] (Fig. 3).
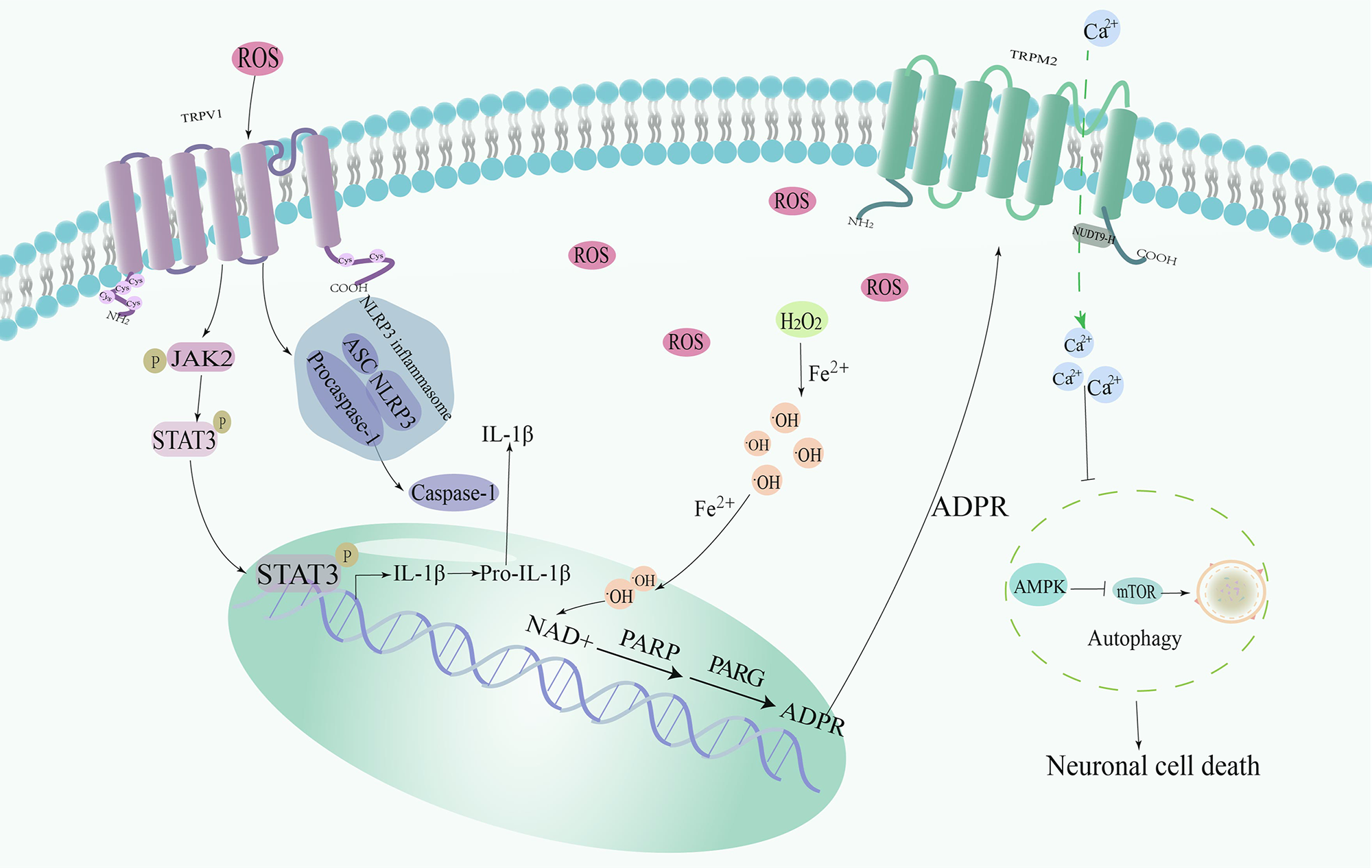 Fig. 3.
Fig. 3.Mechanism of redox-sensitive TRP channels involved in Cerebral
IRI (CIRI). TRPV1 regulates the expression of target genes (glial fibrillary acidic protein and IL-1
Due to the high rate of oxygen consumption by renal cells, the kidneys are
greatly affected by arterial flow obstruction and oxygen shortage. Renal IRI
(RIRI) is a common phenomenon in a wide range of diseases and surgeries. In this
process, the ischemic state of the kidneys is abruptly broken, and subsequent
reperfusion of blood flow leads to cellular damage and death. RIRI is the leading
cause of AKI, a disease with high mortality and morbidity [83]. Macrophages, as
part of the immune response, infiltrate the injured tissue in large numbers [84].
Earlier studies of TRPA1 have focused on its role in pain, inflammation, and
sensory transmission. However, recent studies have found that ablation of TRPA1
triggers the increase and infiltration of classically activated macrophages in
mice, further contributing to increased renal inflammation [60]. In
vivo, a variety of experimental models have been used to study the pathogenic
mechanisms and protective strategies of ischemic AKI, among which the mouse
IR-induced RIRI model is more widely used. In this model, renal tubular
epithelial cells show upregulated expression of TRPA1, which acts as an OS sensor
and promotes the transcription of inflammatory factors by activating the
mitogen-activated protein kinase/nuclear factor kappa B (NF-
Hepatic IRI (HIRI) has been the focus of clinical and basic research, particularly its relationship with OS. Recent studies have revealed a series of interesting findings. The production of ROS increases rapidly during reperfusion and is the first destructive event leading to liver injury. TRPM2, as an important ion channel, is closely related to this process. It has been demonstrated that the activation of ROS-dependent NLRP3 inflammatory vesicles is significantly suppressed in macrophages and monocytes when TRPM2 is knocked down or inhibited [85]. This finding indicates that TRPM2 may be a key regulator in the NLRP3 inflammasome pathway. Further studies also found that downregulation of TRPM2 significantly suppresses the NLRP3 inflammasome pathway to attenuate HIRI [64]. A recent study showed that during HIRI, hepatocytes have significant iron overload. ROS, when reacting with iron ions in the cell, produces lipid peroxidation, which further leads to cell membrane disruption and cell death [65]. RNA sequencing analysis showed that genes related to iron overload are significantly enriched in TRPM2-induced IRI. Specifically, HIRI activates TRPM2. Then intracellular calcium accumulates in the mitochondria throughthe mitochondrial calcium uniporter 1 (MCU1) complex, increasing the expression of arachidonate 12-lipoxygenase, which in turn, promotes lipid peroxidation directly inducing iron overload in hepatocytes and aggravating the injury [66] (Fig. 4).
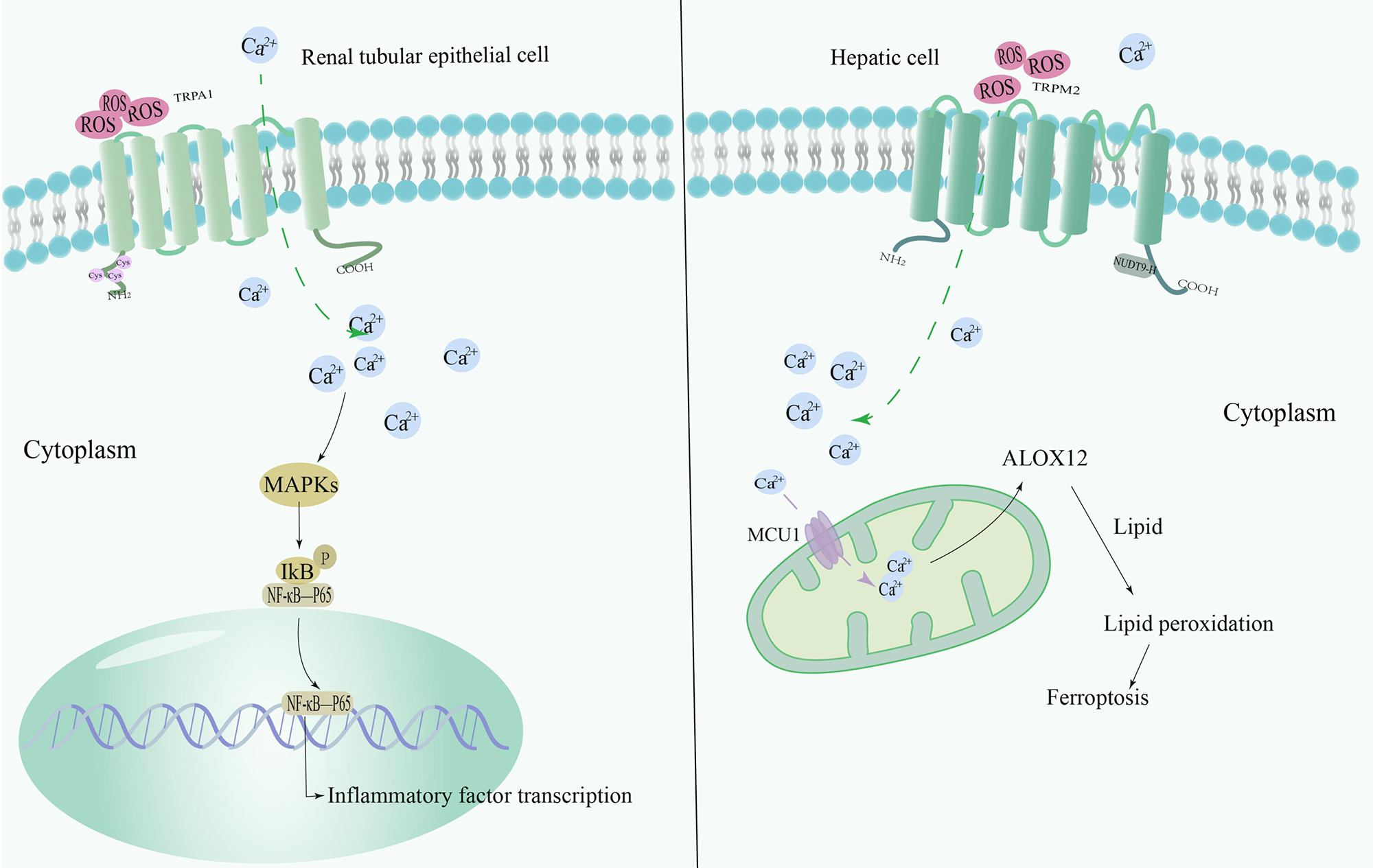 Fig. 4.
Fig. 4.Mechanism of redox-sensitive TRP channels involved in
RIRI/HIRI. TRPA1 is an OS sensor, and in renal tubular epithelial cells, TRPA1
expression is upregulated and promotes the transcription of inflammatory factors
through activation of the MAPK/NF-
TRP channel modulators, a promising avenue in the realm of medical research, have shown remarkable potential in mitigating the detrimental effects of IRI. Since OS and ROS can induce autophagy, this suggests that under certain circumstances, cells may utilize autophagy as a strategy to manage OS. TRPV1 contributes to the maintenance of cellular homeostasis by stimulating autophagy in microglia. This autophagic process has an additional benefit: it inhibits activation of the NLRP3 inflammasome [86]. 6-Gingerol, a phenolic compound extracted from ginger, is expected to be an effective drug for the treatment of IRI by targeting the TRPV1/FAF1 complex and promoting autophagy [77]. Recently, researchers established a model of ROS-mediated cellular OS injury to study the role of TRPV1. The results showed that the TRPV1 inhibitor, iodoresiniferatoxin, antagonized autophagy related gene 5-mediated autophagy, thereby exacerbating oxidative cell damage and apoptosis [75]. Inhibition of TRPA1 during the critical phase of myocardial reperfusion provides significant benefits by limiting calcium overload in cardiomyocytes [68]. Confocal microscopy and electrophysiology have shown that TRPA1 is co-localized with a gap connexin enriched in the intercalation disc [69]. However, the creation of a novel FLP reporter mouse line confirmed that TRPA1 expression is limited to sensory neurons [87]. This exciting development brings us one step closer to understanding the role of TRPs in ischemic disease. Hydra Biosciences identified a selective antagonist of TRPA1, HC-030031 (HC), through high-throughput screening [88]. Although HC reduces hypoxic ischemia-induced brain damage, the combination of HC and desflurane provides similar protection as HC or desflurane alone [89]. Similarly, the cardioprotective benefits of remote preconditioning of trauma were eliminated by the use of the TRPV1 inhibitor capsazepine [90]. By contrast, when TRPV1 in the heart is activated by capsaicin, it effectively reduces ROS production by modulating the influx of calcium into cardiac cells and increasing nitric oxide bioavailability [91]. MIRI is particularly damaging to cardiomyocytes in the context of diabetes. Recent studies have highlighted the therapeutic potential of oleoylethanolamide (OEA), a TRPV1 agonist. OEA activates the phosphoinositide 3-kinase/Akt signaling pathway in these rats, which significantly reduces cardiomyocyte apoptosis [92]. Studies on TRP channel modulators mark an important breakthrough in the field of IRI and provide strong support for future clinical practice and research.
Insights into the role of TRPM2 channels in IRI and the search for ways to inhibit their overactivation have become a hot topic of research. 8-bromo-cyclic ADP-ribose, a cyclic ADPR antagonist, is able to reduce TRPM2 expression and attenuate renal injury [93]. The antidepressant duloxetine was recently found to also inhibit CIRI-induced TRPM2 channel activation and may be an effective treatment for reperfusion injury [57]. A subsequent article on heat-evoked inward currents of TRPM2 noted that the inhibition of duloxetine is significantly concentration-dependent, and the plasma concentration of duloxetine is usually in the nanomolar range. Therefore, the correlation between duloxetine and TRPM2 remains to be verified [94]. Undoubtedly, advances in this field are expected to provide novel therapeutic strategies for the treatment of heart disease, stroke, and other ischemic disorders. Currently, available TRPM2 inhibitors have limitations in terms of selectivity and potency, which make it challenging for researchers, especially in the absence of ideal drug tools. Thus, there is an urgent need to develop novel small-molecule TRPM2 inhibitors that are highly selective, as well as potent enough to effectively intervene in the function of TRPM2 channels [95]. Anthranilic acid (ACA) is a widely used inhibitor for TRPM2 studies, but its inhibitory effects lack specificity. By analyzing the structure of ACA and structurally modifying each part, researchers designed and synthesized 59 novel ACA derivatives to investigate the effects of different structural fragments on the inhibitory activity of TRPM2 channels [96]. Among these novel molecules, compound A23 stands out as a novel TRPM2 inhibitor, exhibiting high selectivity and potency. Bilirubin acts as an endogenous direct activator of TRPM2 channels and this novel gating mechanism can also be blocked by compound A23 [58]. Subsequently, the intervention effects of the TRPM2 small molecule inhibitor A10 have received much attention. Researchers have conducted experiments on animal models. Administration of A10 through the tail vein significantly alleviates the damage caused by hepatic ischemia and reperfusion, demonstrating its great potential for liver protection [66]. These findings provide useful molecular tools for the study of physiological and pathophysiological functions associated with TRPM2 channels, as well as potential clinical therapeutic agents for the treatment of ischemic injury.
The development of nanotechnology has revolutionized drug delivery systems. These nanocarriers are mainly categorized into two main groups, natural polymers and synthetic polymers, most of which possess excellent biocompatibility and stability. They are safe and non-toxic and can be modified in a variety of ways to meet the needs of specific applications [97]. When nanoparticles are used as drug carriers, they significantly enhance the ability of drug molecules to penetrate cell membranes and tissues. This property is particularly important for drugs that have difficulty crossing biological barriers. In addition, the special surface of nanoparticles enables the formation of higher localized concentrations of the drug at the target site, which enhances the absorption efficiency and bioavailability of the drug [98]. In the field of nanomedicine, researchers are continuously seeking innovative solutions to address specific medical challenges. One such breakthrough is the development of hyaluronic acid-coated bilirubin nanoparticles, a novel approach that promises to selectively target injured kidneys to mitigate OS and inflammation-induced damage while inhibiting renal tubular cell apoptosis [9]. NL-1-loaded nanoparticles have the unique ability to selectively target the cytoplasm of brain endothelial cells, making them ideal candidates for drug delivery under CIRI [10]. An innovative study found that by precisely controlling the release of curcumin, laser-irradiated Nanoparticles can cross the blood-brain barrier and affect the release of curcumin, which in turn inhibits the activation of TRPM2 channels [99]. Nanotechnology not only improves therapeutic efficacy but also helps to reduce drug side effects, enabling a more precise and safer drug delivery, and providing new therapeutic strategies for IRI.
Acupuncture, as a traditional Chinese medicine therapy, has been combined with
modern medicine to produce a new therapy, namely electroacupuncture therapy.
Electroacupuncture therapy can provide electrical stimulation to acupuncture
points in the body and is used to activate neuron network, in order to regulate
the function of specific organs and treat various diseases. It is an important
part of the new science of bioelectronic medicine. Electroacupuncture
pretreatment downregulates the expression of TRPV1 in neurons, which not only
promotes the release of antioxidant enzymes such as glutathione and superoxide
dismutase but also inhibits the production of inflammatory factors (IL-
| Modulators | Type | Effect | References |
| 6-Gingerol | TRPV1 inhibitors | A phenolic compound extracted from ginger promotes autophagy. | [77] |
| Iodoresiniferatoxin | TRPV1 inhibitors | Antagonizes autophagy-related gene 5-mediated autophagy, thereby exacerbating oxidative cell damage and apoptosis. | [75] |
| Capsazepine | Antagonist of TRPV1 | Blunting remote preconditioning of trauma-induced cardioprotection via the TRPV1 receptor. | [90] |
| HC-030031 | Antagonist of TRPA1 | Induces neuroprotection and reduces IRI-induced brain tissue damage. | [89] |
| Capsaicin | Agonist of TRPV1 | Activation of TRPV1 counteracts the damage caused by ischemia and reperfusion by modulating the nitric oxide pathway of CGRP. | [91] |
| Oleoylethanolamide | TRPV1 agonist | Activation of TRPV1 regulates the phosphoinositide 3-kinase/Akt signaling pathway and attenuates apoptosis in diabetic rats with myocardial ischemia/reperfusion injury. | [92] |
| 8-bromo-cyclic ADP-ribose | TRPM2 antagonist | Inhibition of RIRI. | [93] |
| Duloxetine | TRPM2 antagonist | Protection against CIRI by TRPM2 inhibition. | [57] |
| A23 | TRPM2 blocker | Attenuation of ischemic brain injury exacerbated by direct bilirubin agonism of TRPM2 channels. | [58] |
| A10 | TRPM2 blocker | Pharmacological inhibition of TRPM2 attenuates hepatic IRI. | [66] |
| Electroacupuncture | Acupuncture | Suppressed TRPV-1 expression and induced neuroprotection in CIRI. | [100, 101] |
Since attention has been drawn to the phenomenon of IRI, this has been a hot topic of research in the medical community. A particular point of interest is the role played by ROS produced by IRI. These reactive molecules have the capacity to activate redox TRPs that play pivotal roles in cellular signaling and tissue damage. The discovery of redox TRPs offers a fresh perspective into the mechanistic understanding of IRI. Regulating the activity of these channels could potentially pave the way for novel therapeutic strategies against IRI. We further delved into the pharmacological potential of modulating redox TRPs for treating IRI, discussing both conventional drug treatments and cutting-edge nanoparticle drug delivery systems. Encouragingly, methods for screening TRP channel modulators have also improved. Based calcium ion imaging and the patch clamp method for direct determination of intracellular calcium ion concentration, Pengfei’s team from Peking University used microfluidic technology to search for more potential TRP regulators, which also provides new insights into the study of therapeutic targets.
MR conceptualized the topic of the paper and completed the writing of the paper. LJY helped to identify the topic of the article and the framework for writing the article. YCT provided advice on the drawing of the image tables. LLS, LF were in charge of collecting the literatures, and participated in the revision and correction of the manuscript. All authors read and approved the final manuscript. All authors participated fully in the work, assumed public responsibility for appropriate portions of the content, and agreed to be accountable for all aspects of the work. All authors contributed to editorial changes in the manuscript.
Not applicable.
Not applicable.
This work was supported by a fund from National Nature Science Foundation of China (No.81974102).
The authors declare no conflict of interest.
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.